(Carregando Índice)... (Carregando Índice)... |
Autores:
Daniele Fricke
Médica neurologista e neurofisiologista do Complexo Hospitalar Santa Casa de Porto
Alegre e do Hospital Moinhos de Vento. Título de Especialista pela Academia Brasileira de Neurologia e pela Sociedade Brasileira de Neurofisiologia Clínica (SBNC). Mestre em Clínica Médica pela UFRGS.
Roberta Diehl Rodriguez
Médica neurologista.
Última revisão: 23/04/2014
Comentários de assinantes: 0
Um paciente do sexo masculino, 63 anos, empresário de uma multinacional, apresentou tremor no membro superior direito há um ano, sem prejuízo funcional. Ele percebeu o tremor à noite enquanto assistia à televisão. Relatou também que seus familiares notaram uma assimetria em seus movimentos durante exercícios de caminhada: o lado direito não se movia como o esquerdo. Os colegas de trabalho reclamaram que ele não era mais tão sorridente quanto antes e parecia estar fugindo dos compromissos sociais. O paciente atribuiu seus sintomas ao excesso de trabalho e ao estresse e afirmou que passaria em seguida.
No ano seguinte, passou a apresentar também tremor na mão esquerda e sentiu que seu corpo estava muito lento, demorando muito mais tempo para fazer suas atividades, inclusive tomar banho. Por isso, procurou auxílio médico. Ele afirmou não haver história familiar de sinais e sintomas semelhantes e também disse não apresentar doenças preexistentes, nem utilizar medicamentos cronicamente.
Ao realizar exame, foram observados expressão facial embotada, marcha com passos curtos, com pouco movimento de balanço dos membros, tremor de repouso no hemicorpo direito e no membro superior esquerdo, movimentos manuais lentos e com amplitude reduzida, rigidez emrodadenteada, sem instabilidade postural. Em relação aos nervos cranianos, à coordenação, à sensibilidade e aos reflexos, não houve particularidades. O paciente realizou ressonância magnética nuclear de encéfalo (RM) e exames laboratoriais gerais, dosagem de vitamina B12, ácido fólico, sorologia para sífilis e hormônio tireoestimulante (TSH), que evidenciaram resultados normais. Por meio de eletroneuro-miografia (ENMG), foi possível confirmar tremor em repouso de 4 a 6 Hz.
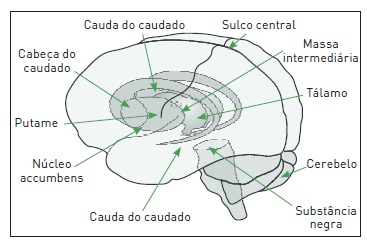
Os distúrbios do movimento compõem um grupo heterogêneo de doenças que alteram o controle voluntário do movimento sem afetar diretamente a força muscular, a sensibilidade ou a função cerebelar. Ocorrem devido a uma disfunção nos núcleos da base (NBs), que são massas subcorticais simétricas localizadas nas porções mais inferiores dos hemisférios cerebrais. Os NBs são constituídos de cinco núcleos interconectados: caudado, putame, globo pálido (GP), substância negra e núcleo subtalâmico. Os dois primeiros, por apresentarem estruturas anatômicas e funcionais semelhantes, são denominados, em conjunto, de corpo estriado (CE) (Fig. 93.1).
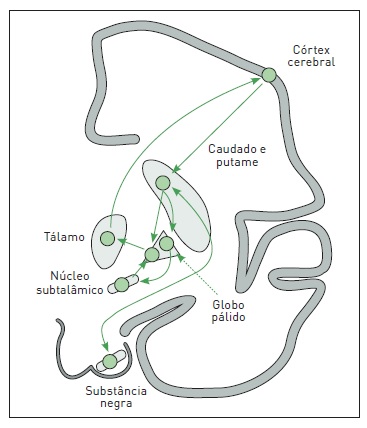
O funcionamento básico dos NBs baseia-se em mecanismos inibitórios e excitatórios distribuídos em três circuitos básicos. O primeiro circuito, corticocortical, inicia no córtex cerebral, passa por caudado e putame, segmento interno do globo pálido, tálamo e volta ao córtex. O segundo, nigroestriatal, conecta a substância negra com o caudado e o putame. E o terceiro, estriatopalidal, projeta-se do caudado e do putame para o segmento externo do globo pálido, núcleo subtalâmico e volta para o segmento interno do globo pálido (Fig. 93.2).

Figura 93.1
Localização anatômica dos núcleos da base.
Fonte: Adaptada de Blandini e colaboradores.1
Figura 93.2
Funcionamento básico dos núcleos da base.
Fonte: Adaptada de Aminoff e colaboradores.2
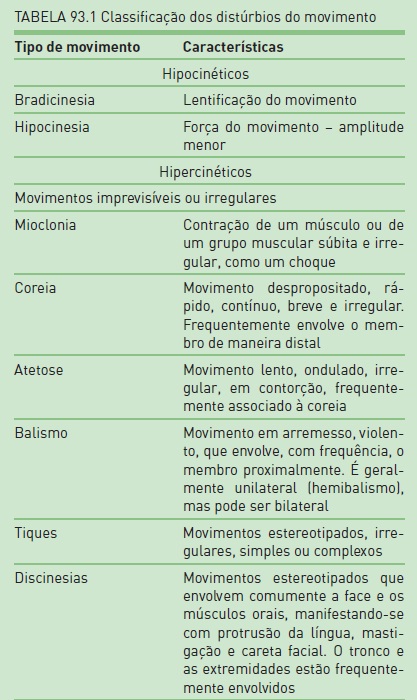
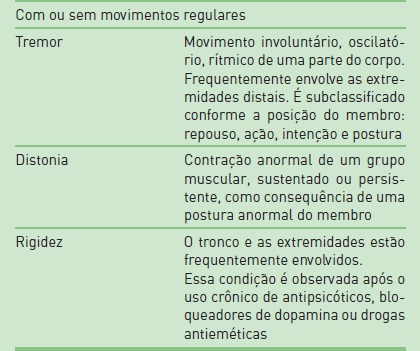
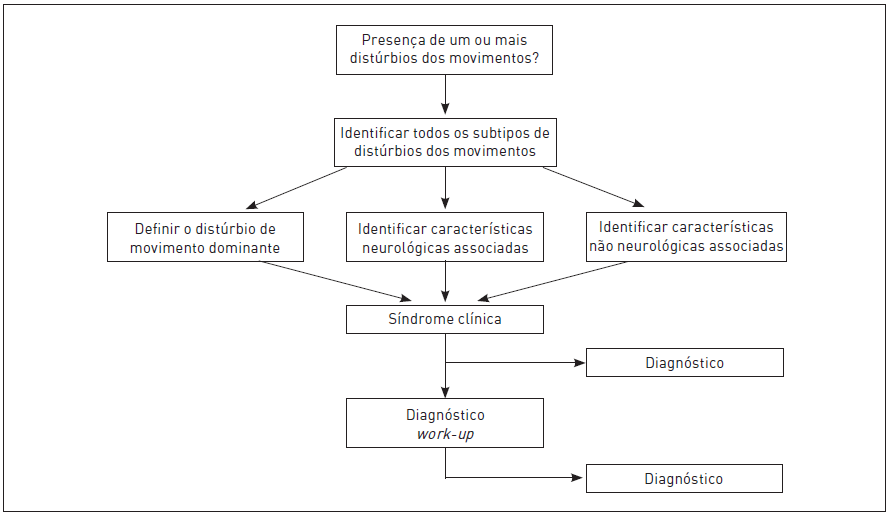
A apresentação clínica dos distúrbios do movimento é complexa e variável, tornando o diagnóstico muitas vezes difícil, mesmo quando realizado por especialistas experientes. A primeira etapa na abordagem diagnóstica é definir a fenomenologia do problema. De um modo geral, pode-se dividir os sinais/sintomas dos distúrbios do movimento em duas grandes categorias: distúrbios hipercinéticos e hipocinéticos (Tab. 93.1).
Definida a fenomenologia, deve-se identificar outros achados neurológicos e não neurológicos e tentar definir se há uma síndrome específica (Fig. 93.3). Para isso, a realização de exames complementares pode ser de grande valia.
A seguir, são apresentadas e definidas algumas síndromes mais prevalentes, caracterizadas por movimentos anormais.
A doença de Parkinson (DP) é neurodegenerativa progressiva, sendo responsável por aproximadamente 80% dos casos de parkinsonismo.
Há degeneração dos neurônios que contêm neuromielina no tronco cerebral, especialmente na camada ventral da parte compacta da substância negra e no locus coeruleus. Muitos dos neurônios sobreviventes contêm inclusões proteináceas citoplasmáticas eosinofílicas designadas corpos de Lewy, a característica patológica típica da doença. Quando os sintomas manifestam-se, a substância negra já perdeu cerca de 60% dos neurônios dopaminérgicos, e o conteúdo de dopamina do estriado é cerca de 80% inferior ao normal.
A etiologia dessa doença é multifatorial: envelhecimento normal, influência genética, fatores ambientais e tóxicos.
A DP afeta entre 100 a 200/100.000 pessoas com mais de 40 anos de idade. Não ocorre comumente em pessoas jovens com menos de 40 anos, e a incidência aumenta rapidamente após os 60 anos, sendo a média da idade de diagnóstico de 70,5 anos. A taxa de incidência é maior em homens (3:2).
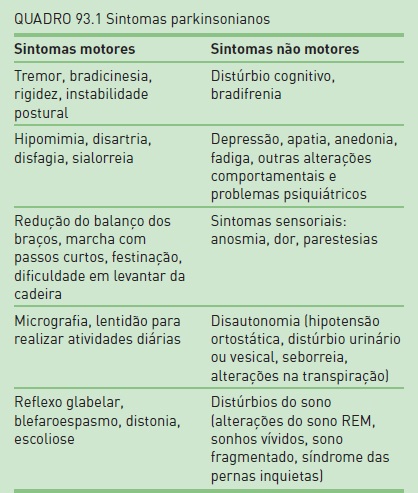
Há quatro sinais cardinais da DP que podem ser agrupados com o acrônimo TRAP: tremor de repouso, rigidez, acinesia (ou bradicinesia) e instabilidade postural. Adicionalmente, existem outros sintomas, como postura fletida e fenômeno de congelamento. O início é insidioso, e o tremor de repouso é o primeiro sintoma observado em 70% dos casos. Os sintomas em geral apresentam-se de maneira unilateral e, em seguida, bilateral devido à progressão da doença.
Além dos sinais motores que definem o parkinsonismo, a maioria dos pacientes com DP também evidencia sinais comportamentais (Quadro 93.1).
A DP, de modo didático, tem duas formas básicas de apresentação: com predomínio de tremor e acinético-rígida.
Figura 93.3
Uma abordagem sistemática para diagnóstico de pacientes que apresentam distúrbios do movimento.
Fonte: Adaptada de Abdo e colaboradores.3
Forma com predomínio de tremor. Apresenta-se como tremor de repouso, unilateral, de início insidioso e piora progressiva, geralmente em meses ou anos, que afeta as demais extremidades e o segmento cervical com o desenvolvimento da doença. É a forma que ocorre na maioria dos casos de DP idiopática.
Forma acinético-rígida. É mais dificilmente reconhecida. Nota-se uma maior lentidão para realizar atividades cotidianas e marcha com passos curtos. Em alguns casos, essa restrição de movimentos é tão acentuada em somente um dos hemicorpos que o paciente é diagnosticado erroneamente com doença cerebrovascular. Esses casos podem passar anos sem o estabelecimento do diagnóstico correto e, em casos extremos, por até uma década.
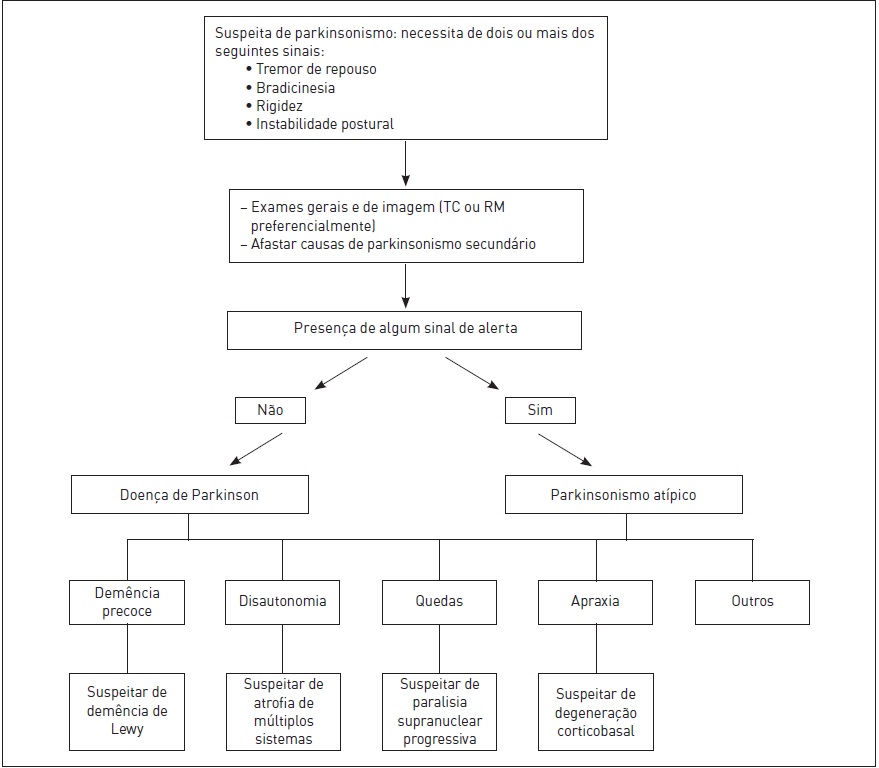
O diagnóstico tem como base critérios clínicos. Não há ainda um marcador biológico específico ou um teste diagnóstico definitivo para a DP. O critério-padrão é a apresentação, na autópsia, de corpúsculos de inclusão de Lewy. Os exames de imagem e laboratoriais são utilizados para descartar outras causas de parkinsonismo. Na prática clínica, o diagnóstico é baseado na existência de uma combinação de sinais motores cardiais (tremor, bradicinesia, rigidez e instabilidade postural) associados, sintomas excludentes e resposta à levodopa. Embora o diagnóstico de DP seja simples em pacientes com uma apresentação clássica, a diferenciação de outras formas de parkinsonismo pode ser um desafio nos estágios iniciais da doença quando os sinais e os sintomas coincidem com os de outras síndromes. Cerca de 5 a 10% dos pacientes com DP são diagnosticados erroneamente, enquanto 20% dos pacientes diagnosticados com DP apresentam outro diagnóstico na autópsia, como paralisia supranuclear progressiva, atrofia de múltiplos sistemas e doença cerebrovascular.
O exame clínico com realização de acompanhamento a longo prazo parece ser o melhor método para a confirmação do diagnóstico no decorrer da vida do paciente. A ocorrência de alguns sinais requer uma avaliação mais detalhada para que sejam descartadas outras causas de parkinsonismo. Deve-se atentar para os seguintes:
•Demência precoce e proeminente
•Alucinações precoces
•Sinais simétricos
•Disfunção bulbar precoce
•Disfunção precoce da marcha
•Redução, no primeiro ano, de sintomas
•Dependência de cadeira de rodas nos primeiros cinco anos
•Falência autonômica precoce
•Apneia do sono
•Apraxia
•Membro alienígena (movimentos involuntários no membro afetado)
•Perda sensitiva cortical
•Outros achados no exame neurológico, como síndrome cerebelar, sinais piramidais e distonia precoce
•Ausência de resposta à levodopa
As alterações parkinsonianas podem ser classificadas em quatro tipos: primário ou idiopático, secundário (adquirido, sintomático), heredodegenerativo, degeneração de múltiplos sistemas. Embora a resposta à levodopa sugira a ocorrência de DP, não diferencia definitivamente a DP de outras doenças parkinsonianas, pois muitas formas de parkinsonismo sintomático (pós-encefalítico, induzido por reserpina) e de síndromes parkinsonianas atípicas respondem à levodopa em seus estágios iniciais (Fig. 93.4).
Em casos de parkinsonismo induzido por drogas, as que bloqueiam os receptores de dopamina D2 no estriado (fenotiazinas, butirofenonas) ou depletam dopamina no estriado (reserpina, tetrabenazina) podem induzir um estado parkinsoniano. Em geral, o parkinsonismo apresenta-se de forma simétrica, com evolução mais branda, e há restabelecimento completo da condição do paciente com a suspensão da droga, mas isso pode demorar algumas semanas ou não ocorrer. A ocorrência de tremor é relativamente incomum, enquanto a bradicinesia em geral é simétrica e a característica neurológica mais evidente. Os sintomas geralmente se desenvolvem em até três meses a partir do início da medicação. O uso de drogas anticolinérgicas pode atenuar os sinais e os sintomas parkinsonianos. A administração de bloqueadores do canal de cálcio com ação antivertiginosa (p. ex., flunarizina) e de neurolépticos (haloperidol) é a mais
Suspeita de parkinsonismo: necessita de dois ou mais dos seguintes sinais:
•Tremor de repouso
•Bradicinesia
•Rigidez
•Instabilidade postural
–Exames gerais e de imagem (TC ou RM preferencialmente)
–Afastar causas de parkinsonismo secundário
Presença de algum sinal de alerta frequente causadora dessa doença no Brasil. As drogas antipsicóticas atípicas, como a clozapina e a quetiapina, são os antipsicóticos com menor probabilidade de induzir ou agravar o parkinsonismo.
Figura 93.4
Abordagem diagnóstica do paciente com parkinsonismo.
Fonte: Adaptada de Maia e Frota.4
Essa é provavelmente a segunda causa mais frequente de demência degenerativa depois da doença de Alzheimer. Clinicamente é definida como uma síndrome demencial progressiva com déficits visuoespaciais e atencionais proeminentes, flutuações na atenção e na cognição, alucinações visuais recorrentes e persistentes, parkinsonismo e sensibilidade a medicações neurolépticas. Não se pode distinguir o parkinsonism, que se manifesta com tremor, rigidez, bradicinesia e perda dos reflexos posturais, da DP. Os corpos de Lewy são observados no córtex cerebral. Cerca de 40% dos pacientes com DP desenvolvem também um quadro demencial, segundo estudos de base populacionais, porém isso ocorre em fases avançadas, com manifestações motoras existentes há alguns anos.
A PSP é uma doença degenerativa multissistêmica que pode ser diferenciada da DP de maneira fácil quando os pacientes apresentam as características clínicas típicas de paralisia supranuclear do olhar, incluindo predominantemente o olhar vertical, parkinsonismo, paralisia pseudobulbar e síndrome do lobo frontal. Nos estágios iniciais, a PSP pode ser confundida com a DP em pacientes sem paralisia do olhar ou quando o parkinsonismo domina o quadro clínico.
Em casos de PSP, sintomas axiais (pescoço e tronco) predominam, e a ocorrência de tremor de repouso é incomum. A bradicinesia ocorre de forma simétrica e pode ser grave. Há instabilidade postural e quedas geralmente no primeiro ano da doença. A marcha apresenta base mais ampla e instabilidade. A expressão facial e a inclinação da cabeça para trás, devido à incapacidade de elevar os olhos de forma voluntária, podem evidenciar uma aparência característica que pode ser reconhecida imediatamente. Os homens são afetados duas vezes mais do que as mulheres, e o distúrbio manifesta-se inicialmente entre 45 e 75 anos.
Embora sejam frequentemente esporádicos, casos familiares de PSP foram relatados. Raramente se obtém melhora dos pacientes que realizam o tratamento dopaminérgico, porém aqueles com PSP com sintomas parkinsonianos proeminentes podem responder de maneira favorável à levodopa. O principal achado neuropatológico é a degeneração neuronal com emaranhados neurofibrilares de proteína Tau em estruturas subcorticais e do tronco cerebral.
Esse tipo de atrofia uma degeneração de múltiplos sistemas esporádica, associada a depósito de alfa-sinucleína no SNC. Ela afeta 10% dos pacientes com parkinsonismo. O espectro patológico integral consiste em perda neuronal e gliose no neoestriado, na substância negra, no globo pálido, no cerebelo, nas olivas inferiores, nos núcleos da base da ponte, nas células do corno intermediolateral, nas células do corno anterior e nos tratos corticoespinais.
A AMS pode apresentar-se como uma forma predominante ou exclusivamente cerebelar (atrofia olivopon-tocerebelar), parkinsoniana (degeneração nigroestriatal) e outra forma predominantemente autonômica (síndrome de Shy-Drager). Pode ser muito difícil diferenciar a variante parkinsoniana da atrofia de múltiplos sistemas (AMS-P) da DP.
A AMS-P ocorre em indivíduos um pouco mais jovens do que os com DP, mas o pico de incidência continua sendo aos 60 anos. A doença pode ser diagnosticada precisamente apenas se sinais proeminentes de falência autonômica, como impotência ou hipotensão postural, desenvolverem-se de maneira precoce no curso da doença ou se houver uma síndrome cerebelar evidente. Além da disautonomia, outros sinais podem possibilitar a diferenciação entre AMS-P e DP, tais como estridor noturno, curso rápido, quedas e instabilidade precoce, sinais de lesão do trato piramidal, disartria grave e insuficiente ou transitória resposta à levodopa.
Degeneração corticobasal. A degeneração corticobasal é um distúrbio degenerativo raro e não familiar. A apresentação inicial da doença é insidiosa e tipicamente unilateral, com rigidez e distonia acentuadas no braço afetado. São também observados sinais corticais de apraxia, fenômeno do membro alienígena, perda sensorial cortical, mioclonia sensitiva a estímulo e mioclonias corticais reflexas do membro afetado. Em pacientes com essa condição, a fala é hesitante, a marcha é deficiente e ocasionalmente se evidencia um tremor de ação. Sinais de liberação frontal, reflexos tendíneos aumentados e respostas plantares extensoras também podem ser verificados nesses casos.
A causa comum de incapacidade é a apraxia e a incoordenação, e não os déficits extrapiramidais. Pode haver semelhança com PSP, e ambas as condições acumulam a proteína Tau. Os sinais motores raramente respondem à levodopa, e a doença passa a ser bilateral e produzir incapacidade grave em 2 a 7 anos. A ocorrência de oftalmoplegia supranuclear é comum inclusive nos estágios mais avançados da doença.
O tratamento de pacientes com DP pode ser farmacológico, cirúrgico e não farmacológico. O objetivo é melhorar a qualidade de vida dos pacientes, reduzir as incapacidades o quanto for possível com a menor dose efetiva de medicação, pois não há tratamento curativo.
– Selegilina: a selegilina é um inibidor da monoaminoxidase B (IMAO-B) que apresenta fraca atividade dopaminérgica. Esse medicamento foi muito utilizado devido ao suposto efeito neuroprotetor, que atualmente está sendo questionado. Os pacientes em fases iniciais geralmente evidenciam resultado satisfatório com essa medicação. Ela apresenta efeito modesto sobre a rigidez e a bradicinesia. Há um novo IMAO-B, rasagilina, que tem sido utilizado como monoterapia inicial para pacientes com DP ou em combinação com levodopa para pacientes com DP mais avançada. Os IMAO-B podem aumentar o risco de neoplasia de pele (melanoma) em pacientes com DP.
– Amantadina: esse fármaco apresenta atividade dopaminérgica satisfatória, além da propriedade de reduzir as discinesias quando elas começam a se manifestar. Pode causar alucinações em pacientes em fases mais avançadas da doença, mas, com a redução da dose, há regressão do sintoma.
– Agonistas dopaminérgicos: os agonistas dopaminérgicos estimulam diretamente os receptores de dopamina. Foi reduzida a administração da bromocriptina antigamente utilizada pela introdução dos agonistas não ergotamínicos, como o pramipexol e o ropinirol. Estes apresentam atividade dopaminérgica semelhante à da levodopa, porém com maior meia-vida, diminuindo as flutuações, e evidenciam também efeito neuroprotetor.
– Anticolinérgicos: o cloridrato de triexifenidil é o mais importante deste grupo. Os anticolinérgicos não são administrados com frequência devido à pouca tolerabilidade e eficácia. Podem ser utilizados por pacientes jovens com tremor incapacitante.
– Levodopa: esse fármaco é utilizado como a principal linha de tratamento da DP, evidenciando alto índice de eficácia em sintomas como bradicinesia e tremor. Em geral, a utilização inicial é adiada ao máximo devido a complicações motoras que ocorrem com o uso a longo prazo. É a primeira opção de medicamento para pacientes com DP de início mais tardio. A levodopa deve ser sempre administrada com inibidor da enzima que a degrade perifericamente (benserazida, carbidopa), a fim de aumentar sua biodisponibilidade no SNC. O tratamento deve ser iniciado com doses pequenas, aumentando-as de forma progressiva conforme a tolerância do paciente. Ao ajustar a dose de levodopa, deve-se observar a existência de complicações motoras, como as discinesias. Se estas ocorrem logo após a tomada da medicação, podem ser secundárias ao pico de levodopa, evidenciando melhora com a redução da dose; se ocorrem pouco antes da próxima dose, pode-se aumentar a dose, reduzir o intervalo entre as administrações ou, ainda, adicionar um inibidor da catecol-O-metiltransferase (COMT). Os efeitos colaterais principais são náuseas, diarreia e hipotensão postural.
– Inibidores da COMT: esses medicamentos potencializam o efeito da levodopa, aumentando a meia-vida desta no SNC e reduzindo as flutuações motoras e as discinesias. A administração do entacapona é mais segura do que a do tolcapona, que, quando utilizado, precisa que seja realizada monitoração das enzimas hepáticas frequentemente.
Como tratamento não farmacológico, pode-se realizar fisioterapia, acompanhamento fonoaudiológico e terapia ocupacional.
A realização de tratamento por meio de talamotomia ou palidotomia com frequência é útil quando os pacientes não apresentam resultado satisfatório com as medidas farmacológicas ou se desenvolvem reações adversas intoleráveis a medicações antiparkinsonismo. A talamotomia é mais eficaz para o tremor, enquanto a palidotomia, para a hipocinesia.
Atualmente tem sido empregada a técnica de estimulação cerebral profunda de alta frequência por meio do implante de um eletrodo no núcleo ventral intermédio do tálamo, na região subtalâmica ou mesmo no globo pálido. Ao contrário da cirurgia ablativa, a estimulação elétrica com alta frequência não lesiona o tecido. Os principais inconvenientes são o custo e a necessidade de trocar as baterias implantadas a cada dois anos.
A primeira etapa para realizar a avaliação de um paciente com tremor é caracterizá-lo (Tab. 93.2).
A forma mais comum é o tremor de ação, que é uma manifestação de várias doenças, sendo a mais prevalente o tremor essencial (0,4 a 6%; a taxa é maior em indivíduos com mais de 65 anos). Esse tremor, de 4 a 12 Hz, afeta principalmente as mãos, mas também pode comprometer a cabeça, a voz, o tronco e as pernas.
O tremor de ação parece ocorrer devido à disfunção nos circuitos neuronais que passam do cerebelo ao córtex cerebral através do núcleo intermédio ventral do tálamo. Alguns estudos evidenciaram diminuição das células de Purkinje no córtex cerebelar e corpos de Lewy nos locus coeruelus. Parece haver um padrão de herança mendeliana.
O diagnóstico desse tipo de tremor é essencialmente clínico. Uma história detalhada (idade de início, tipo, taxa de progressão, história familiar, existência de outras anormalidades do movimento ou sinais/sintomas de doenças clínicas) auxilia no diagnóstico diferencial:
•Tremor fisiológico exacerbado (por cafeína, cigarro, lítio, prednisona, levotiroxina, adrenérgicos, broncodilatadores, valproato e inibidores seletivos da recaptação da serotonina)
•Doença de Parkinson
•Distonia idiopática de início na vida adulta
•Doença de Wilson
O tratamento deve ser realizado apenas por pacientes que apresentem prejuízo funcional em decorrência do tremor. Tanto a administração de propranolol (160 a 320 mg/dia) quanto a de primidona (65 a 1.000 mg/dia) evidencia resultados efetivos. A cirugia (deep-brain stimulation ou talamotomia) está indicada para os casos graves que não apresentam eficácia com o uso de medicamentos.
A doença de Wilson (DW), ou degeneração hepatolenticular, é uma doença autossômica recessiva do metabolismo do cobre que resulta em cirrose e neurodegeneração.
Essa doença ocorre em indivíduos em qualquer região do mundo, sendo mais comum entre 10 e 30 anos e em filhos de pais consanguíneos. Sua prevalência é de 1/100.000 pessoas, manifestando-se igualmente entre os sexos.
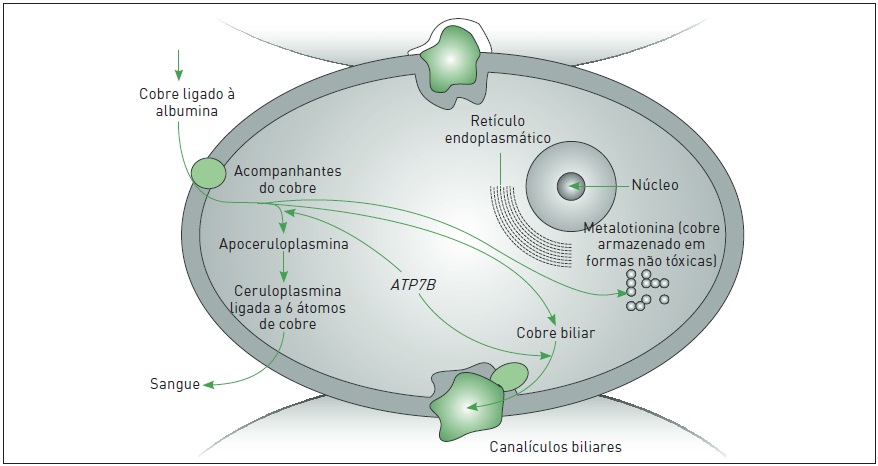
O cobre ingerido na dieta é absorvido pelas células intestinais e armazenado como metalotioneína (uma forma não tóxica). Posteriormente, o cobre entra na corrente sanguínea através de uma proteína transportadora (ATP7A) localizada na membrana do enterócito e liga-se à albumina para chegar ao hepatócito. Dentro dessa célula, o cobre liga-se à metalotioneína através da proteína ATOX1 para armazenamento, e o restante é excretado nos canalículos biliares regulados pela enzima ATP7B. Essa enzima também possibilita a transferência do cobre para apoceruloplasmina a fim de formar a ceruloplasmina (proteína ligada a seis moléculas de cobre). A ceruloplasmina é liberada na corrente sanguínea (carrega 90% do cobre existente no plasma) e age como uma fonte de cobre para os órgãos periféricos, como o cérebro e o rim. A mutação no gene ATP7B resulta em redução da conversão de apoceruloplasmina em ceruloplasmina e uma falência na excreção do cobre nos canalículos biliares. O excesso de cobre nos hepatócitos causa lesão mitocondrial, lesão oxidativa celular e saída de cobre para a corrente sanguínea com consequente sobrecarga para os outros órgãos (Fig. 93.5).
A apresentação clínica é variada e depende do acúmulo de cobre nos órgãos afetados, como se verifica a seguir:
Fígado. Em todos os pacientes, o cobre inicialmente se deposita no fígado, ocasionando inflamação aguda ou crônica com desenvolvimento de cirrose. Sintomas decorrentes das alterações hepáticas incluem fadiga, anorexia, perda de peso, fraqueza generalizada, ascite, icterícia e hepatoesplenomegalia.
Figura 93.5
Representação esquemática do metabolismo do cobre dentro da célula hepática ATP7B, gene da Doença de Wilson.
Fonte: Adaptada de Das e Ray.5
Cérebro. No cérebro, o acúmulo de cobre causa alterações cognitivas, demência, tremor, distonias, bradicinesia, ataxia, disartria, disfonia e disfagia.
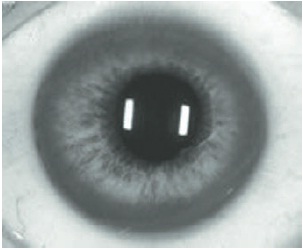
Olhos. O cobre também pode se depositar na córnea, tendo como consequência a formação de um anel de cor marrom-ouro que pode ser observado por meio de exame clínico (Fig. 93.6).
Outros. Alterações psiquiátricas (irritabilidade, agitação, oscilações do humor, ansiedade, histeria, comportamen tos bizarros, depressão), osteoarticulares, hematológicas, renais e cardíacas também são observadas.
Figura 93.6
A liberação aguda de cobre no sangue também causa hemólise.
O diagnóstico precoce é fundamental, pois as alterações hepáticas ocorrem antes mesmo dos sintomas clínicos. É recomendada investigação em todos os pacientes com lesão hepática sem causa aparente ou com sinais/ sintomas extrapiramidais associados a história familiar ou familiares com alteração hepática e neurológica semelhante.
–Laboratorial: nível sérico de ceruloplasmina baixo (< 200 mg/dL); alto nível sérico de cobre livre (> 100 g/ dL); cobre urinário de 24 horas alto (> 100 g).
–Presença de anéis de Kayser-Fleischer (anéis amarelo-ouro ou marrom-esverdeados em torno da córnea dos olhos). As vezes é necessário realizar avaliação por meio da lâmpada de fenda.
–Evidência de nível elevado de cobre no fígado (> 250g/g de peso seco) por meio de biópsia.
–Análise genética: mutações no gene ATP7B (não é uma análise prática, pois mais de 300 mutações já foram identificadas).
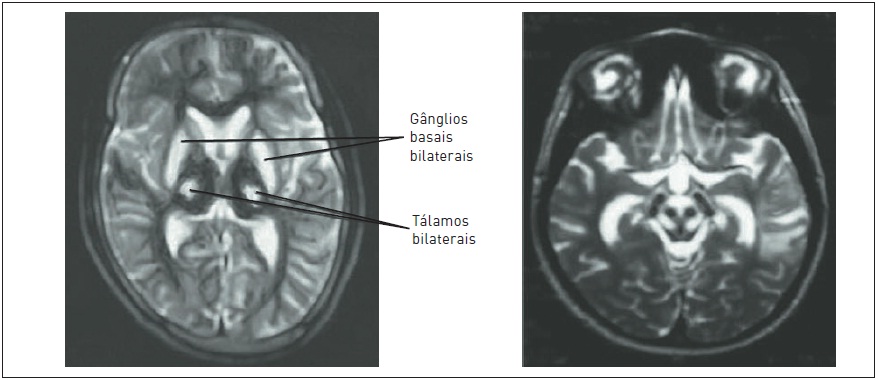
–Imagem: ressonância magnética (RM) do encéfalo com hiperintensidade T2 nos núcleos da base e no tálamo e no mesencéfalo (aspecto de face de panda gigante). RM com espectroscopia evidencia um índice de Nacetilaspartato (NAA): creatina e colina: creatina reduzido (Fig. 93.7A e B).
O objetivo principal do tratamento é remover o excesso de cobre do organismo e prevenir seu acúmulo e deposição. A eficácia do tratamento é avaliada por meio dos níveis séricos de cobre livre e sua excreção urinária. A terapia medicamentosa é realizada com agentes queladores do cobre (D-penicilamina), trientina e acetato de zinco (bloqueia a absorção intestinal) e deve ser continuada durante toda a vida. Alguns alimentos que contêm cobre devem ser evitados, tais como cacau, chocolate, fígado, cogumelos, nozes. O tratamento inadequado pode resultar em lesões irreversíveis em órgãos-alvo. Às vezes é necessária a realização de transplante de fígado.
A hidrocefaleia com pressão normal (HPN) é uma síndrome que apresenta diversas manifestações clínicas, neurológicas e fisiológicas. Clinicamente se evidencia a tríade clássica de distúrbio da marcha, demência e incontinência urinária, com pressão do líquido cerebrospinal normal e resposta à derivação ventricular.
Figura 93.7
Ressonância nuclear magnética ponderada em T2 evidenciando: (A) Hiperintensidade de sinal nos núcleos da base e no tálamo. (B) O típico aspecto de “face de panda gigante” no mesencéfalo.
Fonte: Adaptada de Das e Ray.5
A HPN é uma doença relativamente infrequente, mas que tem recebido bastante atenção devido ao caráter de reversibilidade do quadro demencial. A incidência de HPN nos pacientes com demência varia de 1 a 11% dependendo da amostra populacional.
Acredita-se que a HPN ocorra devido a um distúrbio da dinâmica do líquido cerebrospinal. O líquido cerebrospinal geralmente é produzido pelo plexo coroide, que é formado por tecido secretório especializado localizado nos ventrículos laterais. Ele flui do ventrículo lateral e do terceiro ventrículo através do aqueduto cerebral e do quarto ventrículo e sai do sistema ventricular por dois forames localizados lateralmente: Magendie e Luschka. Então, o líquido cerebrospinal entra e circula através do espaço subaracnóideo em torno do cérebro e da medula espinal. Finalmente ele é reabsorvido pelas granulações aracnoides para a circulação venosa.
Em casos de hidrocefalia, um obstáculo, situado nos espaços meníngeos da base, impede o líquido cerebrospinal de alcançar os locais de reabsorção transependimária, que se estende sobre a substância branca periventricular. O caráter indolente do processo gera um aumento dos ventrículos laterais, com incrementos relativamente pequenos na pressão do líquido cerebrospinal.
O registro contínuo da pressão intraventricular evidencia variações intermitentes, deixando de ser normal durante todo o tempo, o que leva a considerar, como designação mais apropriada, hidrocefalia de pressão intermitente. Essas flutuações periódicas poderiam ser responsáveis pela deterioração progressiva do quadro clínico. Muitos autores questionam a utilização do termo demência curável e demência reversível para a síndrome, pois, apesar de haver grande melhora do quadro com o tratamento, quase sempre déficits cognitivos permanecem.
Sabe-se de uma causa para essa condição na metade dos casos: hemorragia meníngea, meningite ou intervenção cirúrgica que comprometeu a permeabilidade meníngea. Na outra metade dos casos, ainda não se sabe sobre o caráter do processo que causa a doença.
A HPN caracteriza-se tipicamente pelo desenvolvimento gradual (ao longo de semanas a meses) de alterações de marcha, associado a graus variáveis de declínio intelectual, que progridem para níveis mais avançados de demência e incontinência urinária. A tríade de sintomas não ocorre em todos os casos e não estabelece o diagnóstico da doença.
O distúrbio inicial mais frequente é o da marcha. Geralmente há alentecimento da marcha global com tendência à queda e à perda dos movimentos associados. Os pacientes em geral relatam tontura e desequilíbrio. O distúrbio da marcha assemelha-se ao do de parkinsonismo, com passos curtos, arrastando os pés, perda dos reflexos posturais e, às vezes, congelamento. A ocorrência de tremor é rara.
Os sintomas urinários podem manifestar-se como urgência urinária e incontinência urinária franca. A demência é caracterizada por proeminente perda de memória. Havendo sinais corticais, como afasia ou agnosia, deve-se suspeitar de um diagnóstico alternativo, como demência de Alzheimer e demência vascular. Além dos achados típicos, a sintomatologia clínica pode ocorrer concomitantemente a sinais de síndrome frontal: lentidão psicomotora, indiferença afetiva, desinteresse, perturbações amnésicas ou mesmo desinibição comportamental.
Há controvérsias quanto ao melhor critério diagnóstico para identificar os pacientes com HPN. Os testes laboratoriais não apresentam muita utilidade para o diagnóstico, mas são utilizados para descartar a ocorrência de outras etiologias.
A RM é mais específica do que a tomografia computadorizada (TC), mas uma TC normal possibilita a exclusão do diagnóstico. Os achados na TC e na RM são dilatação ventricular, que frequentemente é simétrica e desproporcional à atrofia cortical, edema periventricular com migração de fluidos através das paredes ventriculares, que pode ser melhor avaliado pela RM.
A cisternografia com radioisótopos, em pacientes com HPN, evidencia um retardo na absorção do líquido cerebrospinal sobre a convexidade. Esse exame apresentou evidências no diagnóstico da síndrome, mas não é mais utilizado devido ao risco de complicações e à imprecisão da prova. Atualmente acredita-se que esse método seja superado pela RM, que possibilita também a mensuração da dinâmica do líquido cerebrospinal.
O teste de drenagem denominado tap test é considerado positivo quando há melhora clínica após a drenagem de 40 a 50 mL de líquido cerebrospinal, porém pode apresentar resultado falso-negativo. Há uma tendência atual de se considerar apenas quando o resultado do teste for positivo, isto é, se for negativo, não contraindica a cirurgia. A repetição do teste em geral não apresenta resultados muito precisos. A pressão do líquido cerebrospinal é frequentemente superior à normal, e as concentrações de proteína, glicose e a citometria do líquido cerebrospinal são normais.
A derivação do líquido cerebrospinal é o método-padrão de tratamento. A derivação ventriculoperitoneal é a melhor forma de tratamento, mas não está isenta de complicações, tais como hematoma subdural ou higroma e infecções. Entre as complicações a longo prazo, estão as infecções (8%), a oclusão da ponta do cateter no ventrículo (5%), a drenagem insuficiente (3%) e a drenagem excessiva (3%) e o deslocamento do cateter (2%).
As discinesia tardia (DT) caracteriza-se por movimentos involuntários, não intencionais e repetitivos da língua, dos lábios, da face, do tronco e das extremidades que ocorrem em pacientes que utilizaram cronicamente antagonistas dopaminérgicos. Esses movimentos geralmente não se manifestam durante o sono.
A DT afeta, pelo menos, 20% dos indivíduos que utilizam neurolépticos, com taxas de incidência para novos casos de aproximadamente 3 a 5% ao ano. Essa incidência parece ocorrer de maneira cumulativa e chegar a 30% em idosos expostos ao uso crônico de neurolépticos.
Além das drogas neurolépticas, outros fatores que têm sido relacionados ao aparecimento e ao prognóstico da discinesia tardia são idade, sexo feminino, comorbidade psiquiátrica, existência de outros distúrbios extrapiramidais na fase aguda do tratamento com neurolépticos e diabetes.
A patogênese da DT ainda não está totalmente esclarecida. Existem teorias que defendem a hipersensibilidade dos receptores dopaminérgicos estriatais, e outras que defendem a existência de anormalidades estruturais envolvendo os neurônios gabaérgicos. Nenhum achado patológico é específico para DT.
Não existe um teste específico para identificar um paciente com DT, e o diagnóstico baseia-se na história detalhada e na observação clínica.
O melhor tratamento para pacientes com DT parece ser a prevenção, seja reduzindo a dose da medicação envolvida ou trocando-a (escolher antipsicóticos atípicos). A tetrabenzina, medicação que reduz o nível de dopamina, tem sido utilizada para atenuar alguns sintomas, assim como o pramipexol (agonista dopaminérgico), os benzodiazepínicos e os antagonistas adrenérgicos.
Mesmo com a suspensão do medicamento, os sintomas podem permanecer.
Se o diagrama apresentado na Figura 93.3 for aplicado ao caso em questão, pode-se definir que o paciente evidencia uma alteração do movimento inicialmente restrita a um tremor. A partir de uma avaliação mais detalhada, verifica-se que, além do tremor de repouso, assimétrico a Hz, o paciente também apresenta bradicinesia, hipocinesia e rigidez, ou seja, uma síndrome parkinsoniana.
É iniciado tratamento com levodopa (250 mg) e carbidopa (25 mg), 1 ou 2 comprimidos, três vezes ao dia, em períodos distantes das refeições, com resposta parcial. O aumento da dose para cinco vezes ao dia evidencia melhora significativa.
Os exames complementares com resultados negativos para outras doenças que cursam com parkinsonismo, associados à resposta clínica do paciente à levodopa, confirmam o diagnóstico de doença de Parkinson.
1.Blandini F, Nappi G, Tassorelli C, Martignoni E. Functional changes of the basal ganglia circuitry in Parkinson’s disease. Prog Neurobiol. 2000;62(1):63-88.
2.Aminoff MJ, Greenberg D, Simon RR. Clinical neurology. 6th ed. New York: McGraw-Hill; 2005.
3.Abdo WF, van de Warrenburg BP, Burn DJ, Quinn NP, Bloem BR. The clinical approach to movement disorders. Nat Rev Neurol. 2010;6(1):29-37.
4.Maia FM, Frota NA. Doença de Parkinson [Internet]. Porto Alegre: MedicinaNET; 2008 [capturado em 15 set. 2012]. Disponível em: http:// www.medicinanet.com.br/conteudos/revisoes/1217/doenca_de_parkinson.htm. Acesso restrito.
5.Das SK, Ray K. Wilson’s disease: an update. Nat Clin Pract Neurol. 2006;2(9):482-93.
Brooks DJ. Diagnosis and management of atypical parkinsonian syndromes. J Neurol Neurosurg Psychiatry. 2002;72 Suppl 1:I10-I16.
Celik M, Barkut IK, Oncel C, Forta H. Involuntary movements associated with vitamin B12 deficiency. Parkinsonism Relat Disord. 2003;10(1):55-7.
Colosimo C, Suppa A, Fabbrini G, Bologna M, Berardelli A. Craniocervical dystonia: clinical and pathophysiological features. Eur J Neurol. 2010;17 Suppl 1:15-21.
Czosnyka M, Richards HK, Czosnyka Z. Normal-pressure hydrocephalus. J Neurosurg. 2004;101(6):1083-4.
Fahn S, Przedborski S. Parkinsonismo. In: Rowland LP, organizador. Merrit: tratado de neurologia. 11. ed. Rio de Janeiro: Guanabara Koogan; 2007. p. 769-90.
Jankovic J, Lang AE. Movement disorders: diagnosis and assessment. In: Bradley WG, Daroff RB, Fenichel GM, Jankovic J, editors. Neurology in clinical practice. 4th ed. Philadelphia: Butterworth-Heinemann; 2004. p. 293-322.
Jankovic J. Parkinson´s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry. 2008;79(4):368-76.
Kumar S. Vitamin B12 deficiency presenting with na acute reversible extrapyramidal syndrome. Neurol India. 2004;52(4):507-9.
Litvan I, Bhatia KP, Burn DJ, Goetz CG, Lang AE, McKeith I, et al. Movement Disorders Society Scientific Issues Committee report: SIC Task Force appraisal of clinical diagnostic criteria for Parkinsonian disorders. Mov Disord. 2003;18(5):467-86.
Lorincz MT. Neurologic Wilson’s disease. Ann N Y Acad Sci. 2010;1184:173-87.
Louis ED. Essential tremor. N Engl J Med. 2001;345(12):887-91.
Mattei TA, Aguiar PH, Mattei JÁ. Tendências atuais no diagnóstico e terapêutica da hidrocefalia de pressão normal. J Bras Neurocirurg. 2005;16(1):20-4.
Melo P. Hidrocefalia de pressão normal. In: Melo-Souza SE, editor. Tratamento das doenças neurológicas. Rio de Janeiro: Guanabara Koogan; 2008. p. 316-7.
Patten J. Diagnóstico diferencial em Neurologia. 2. ed. Rio de Janeiro: Revinter; 2000.
Pourmand R. Practicing neurology: what you need to know, what you need to do. 2nd ed. Totowa: Humana; 2008. p. 137-44.
Prockop LD. Hidrocefalia. In: Rowland LP, organizador. Merrit: tratado de neurologia. 11. ed. Rio de Janeiro: Guanabara Koogan; 2007. p. 32532.
Pujari S, Kharkar S, Metellus P. Normal pressure hydrocephalus: longterm outcome after shunt surgery. J Neurol Neurosurg Psychiatry. 2008;79(11):1282-6.
Ropper AH, Brown RH. Adams and Victor´s: principles of neurology. 8th ed. New York: McGraw-Hill; 2005.
Shyambabu C, Sinha S, Taly AB, Vijayan J, Kovoor JM. Serum vitamin B12 deficiency and hyperhomocystinemia: a reversible cause of acute
chorea, cerebellar ataxia in an adult with cerebral ischemia. J Neurol Sci. 2008;273(1-2):152-4.
Soares-Weiser K, Fernandez HH. Tardive dyskinesia. Semin Neurol. 2007; 27(2):159-69
Suchowersky O, Reich S, Perlmutter J, Zesiewicz T, Gronseth G, Weiner WJ. Practice Parameter: diagnosis and prognosis of new onset Parkinson disease (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2006;66(7):968-75.
Tarkowski E, Tullberg M, Fredman P, Wikkelsö C. Normal pressure hydrocephalus triggers intrathecal production of TNF-alpha. Neurobiol Aging. 2003;24(5):707-14.
Tolosa E, Wenning G, Poewe W. The diagnosis of Parkinson´s disease. Lancet Neurol. 2006;5(1):75-86.
Toyama C. Hidrocefalia In: Leite CC, Amaro Jr E, Lucato LT. Neurorradiologia: diagnóstico por imagem das alterações encefálica. Rio de Janeiro: Guanabara Koogan; 2008. p. 384-401.
Tsakanikas D, Relkin N. Normal pressure hydrocephalus Semin Neurol. 2007;27(1):58-65.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.