(Carregando Índice)... (Carregando Índice)... |
Autores:
Daniela Dias Morales
Acadêmica do Curso de Medicina da Universidade Católica de Pelotas (UCPel), RS.
Elvino Barros
Médico nefrologista. Professor associado da Faculdade de Medicina da UFRGS. Doutor em
Nefrologia pela Universidade Federal de São Paulo (UNIFESP).
Francisco José Veríssimo Veronese
Médico nefrologista do Serviço de Nefrologia do HCPA. Professor adjunto do Departamento de Medicina Interna da Faculdade Medicina da UFRGS.
José Vanildo Morales
Professor associado do Departamento de Medicina Interna da Faculdade de Medicina da UFRGS. Doutor em Ciências Médicas: Nefrologia
pela UFRGS.
Última revisão: 05/08/2019
Comentários de assinantes: 0
Uma paciente do sexo feminino, 62 anos, branca, apresentou febrícula há alguns meses, perda de peso e fadiga. Há um mês iniciou um quadro de anorexia e, na última semana, hemoptise por três vezes. Nos últimos dois dias, observou diminuição da diurese e teve episódios de náuseas e vômitos. A pressão arterial foi de 180/100 mmHg. Na história, relatou dois episódios de sinusite no último ano e afirmou não apresentar hipertensão arterial. Com esse quadro clínico, procurou auxílio em uma unidade de emergência. Ao realizar exame físico, foram verificados hipertensão (pressão arterial: 210/120 mmHg), temperatura axilar de 37,8oC, palidez e discreto edema de membros inferiores. Na ausculta pulmonar, foram evidenciados estertores crepitantes em bases. O exame de fundo de olho foi normal. Não havia lesões de pele. Os exames de laboratório realizados e seus respectivos resultados foram os seguintes: hematócrito de 26%, hemoglobina de 8,1 g/dL, ureia de 270 mg/dL, creatinina de 9,5 mg/dL, potássio de 6,8 mEq/L e albumina de 3,5 g/dL. No exame qualitativo de urina, observaram-se: proteínas: ++; hemácias de 321/µL, leucócitos de 98/µL, cilindros granulosos e 2 cilindros hemáticos. O índice proteína total/creatinina em amostra de urina foi de 1,9. A investigação evidenciou fator antinuclear (FAN): não reagente, C3: 115, C4: 32, anti-HIV, anti-HCV e HbsAg negativos, crioglobulinas: negativo, c-ANCA (citoplasmático): positivo em título de 1:320, anticorpo anti-MBG: negativo, eletroforese sérica e urinária sem pico monoclonal. A partir do raio X de tórax foi possível constatar infiltrado intersticial bilateral, do tipo “vidro esmerilhado”, e, da ultrassonografia de vias urinárias, rins de forma e tamanho normais, mas com aumento da ecogenicidade.
O termo “vasculite sistêmica” aplica-se a diversas condições que podem comprometer qualquer tipo de vaso por um infiltrado inflamatório com necrose. Outras denominações utilizadas são angeíte ou arterite. As vasculites são definidas pelo tipo de vaso comprometido, pelo padrão de envolvimento e por testes laboratoriais.
Os estudos sobre os anticorpos anticitoplasma de neutrófilos (ANCA) possibilitaram uma nova abordagem sobre o diagnóstico, a classificação e a evolução clínica. A vasculite renal é a manifestação mais comum e grave das vasculites associadas a ANCA (VAA), ocorrendo em mais de 50% dos casos.
O objetivo deste capítulo é abordar as vasculites sistêmicas e mais especificamente as vasculites de pequenos vasos pauci-imunes associadas a ANCA, incluindo a classificação, a patogênese, as manifestações clínicas, a histopatologia e o manejo terapêutico, de forma atualizada e abrangente.
No Brasil, não existem dados sobre a incidência das vasculites sistêmicas. Na Europa, a incidência anual por milhão da população (pmp) é de 10 a 20 pmp/ano, e a prevalência, de 150 a 200 pmp.1 A incidência aumenta com a idade, tendo um pico entre 65 e 74 anos.2 Ainda não se sabe se existe diferença de incidência entre a poliangeíte microscópica (PAM) e a granulomatose de Wegener (GW). É provável que a ocorrência de PAM seja mais comum no sul da Europa e de GW no norte europeu, mas o registro alemão não evidenciou essa diferença.3
As manifestações clínicas ocorrem fundamentalmente conforme o tamanho e o local do vaso comprometido, podendo apresentar uma forma localizada ou sistêmica, algumas vezes benigna e autolimitada, mas em geral com um comportamento agressivo. Por exemplo, em casos de arterite de células gigantes, as manifestações clínicas mais frequentes são cefaleia, cegueira, surdez, claudicação e redução dos pulsos periféricos.4
Os sinais e sintomas são variáveis e dependem dos vasos acometidos. Em geral, há cefaleia, febre, rash eritematoso polimorfo, eritema da mucosa orofaríngea, eritema da palma da mão e planta dos pés, conjuntivite, edema duro e descamação de extremidades.
As vasculites podem ser classificadas conforme a etiologia ou o tamanho dos vasos comprometidos. A classificação etiológica foi proposta por Lie,5 em 1991, e a classificação de acordo com o tamanho do vaso, a partir de um consenso proposto pelo grupo da Universidade de Chapel Hill.4 A classificação pelo tamanho dos vasos considera somente as vasculites sistêmicas idiopáticas. Essas duas classificações são apresentadas nos Quadros 87.1 e 87.2. Além dessas, existem outras classificações derivadas de outros grupos, como o European Vasculitis Study Group (EUVAS), o European League Against Rheumatism (EULAR) e o Vasculitis Clinical Research Consortium (VCRC).
As vasculites de vasos grandes afetam a aorta e seus ramos principais, como artérias das extremidades, cabeça e pescoço.4,6 Na fase aguda, ocorre uma inflamação granulomatosa e, na fase crônica, é estabelecida uma esclerose vascular extensa com pouca ou nenhuma atividade inflamatória. O processo de esclerose e inflamação causam estreitamento desses vasos e consequentemente isquemia e suas manifestações.
A arterite de células gigantes compromete a aorta e seus ramos principais, com preferência para os ramos extracranianos da carótida e envolvimento frequente da artéria temporal. Essa condição ocorre mais comumente em indivíduos com mais de 50 anos. As manifestações clínicas mais frequentes são cefaleia, comprometimento do osso maxilar, cegueira, surdez, claudicação e redução dos pulsos periféricos. O termo “arterite temporal” não é adequado, pois nem todos os pacientes apresentam comprometimento dessa artéria, e outros tipos de vasculites podem afetar a artéria temporal.4

60
A arterite de Takayasu, além do envolvimento da aorta e de seus ramos principais, pode comprometer a artéria pulmonar. Esse tipo de arterite ocorre mais comumente na Ásia, sendo rara em pacientes com mais de 40 anos. Do ponto de vista clínico, apresenta-se com redução de pulsos, claudicação e hipertensão renovascular.
Deve-se suspeitar de arterite de Takayasu se houver ocorrência de hipertensão renovascular em crianças e adultos jovens. Em pacientes com mais de 50 anos, o envolvimento da artéria renal por aterosclerose é a causa mais comum de estenose.
Em casos de vasculites de vasos médios há comprometimento principalmente das artérias que suprem as vísceras maiores. Nos rins, são afetadas as artérias interlobares e arciformes e, com menos frequência, a artéria renal e as interlobulares. Na fase aguda, há inflamação da parede vascular, com focos de necrose fibrinoide, infiltração com neutrófilos e comumente formação de pseudoaneurismas. As principais complicações são trombose, infarto e hemorragias viscerais.
Em poucos dias, o processo inflamatório diminui, iniciando a fase crônica, com predominância dos leucócitos mononucleares e cicatrização progressiva. Esse tipo de vasculite não causa glomerulonefrite, embora os pacientes possam apresentar hematúria e proteinúria, em geral menor do que 2 g por dia.
A poliarterite nodosa (PAN) atualmente é considerada uma entidade distinta das vasculites microscópicas.
O conceito atual de PAN é uma inflamação necrotizante de vasos médios e de pequenas artérias, sem glomerulonefrite ou vasculite em arteríolas, capilares ou vênulas.
Por esse critério, a ocorrência de glomerulonefrite e/ ou comprometimento de capilar alveolar com hemorragia pulmonar descarta o diagnóstico de PAN e indica a existência de doenças com comprometimento de pequenos vasos. A neuropatia periférica, entretanto, não discrimina PAN e vasculites de pequenos vasos.7
Sob o ponto de vista patológico, não é possível distinguir doença de Kawasaki de PAN. A doença de Kawasaki ocorre de forma aguda em crianças, caracterizada pela síndrome dos linfonodos mucocutâneos, com linfadenopatia não supurativa, febre, rush eritematoso polimorfo, eritema da mucosa orofaríngea, eritema da palma das mãos e planta dos pés, conjuntivite, edema duro e descamação de extremidades. A principal causa de mortalidade é o desenvolvimento de arterite necrotizante, principalmente em artérias coronárias. A existência ou não de linfonodos mucocutâneos é uma forma mais acurada de discriminar doença de Kawasaki de PAN.8
Embora as vasculites de grandes e médios vasos possam afetar os rins, em geral por isquemia, o envolvimento renal é particularmente comum nas várias formas de vasculites de pequenos vasos. Esse tipo de doença pode ser definida como vasculite que compromete vasos menores (arteríolas, vênulas e capilares), mas eventualmente pode envolver algumas artérias. Sob o ponto de vista patológico, há um processo inflamatório com necrose que pode acometer um órgão de forma isolada ou vários órgãos simultaneamente.9-11 De acordo com os padrões observados na imuofluorescêcia, podem-se verificar os seguintes tipos de vasculite:
•Vasculites sem depósitos ou depósitos mínimos, pauci-imunes: poliangeíte microscópica, granulomatose de Wegener, síndrome de Churg-Strauss e vasculite limitada ao rim.
•Vasculites com depósitos de imunocomplexos: púrpura de Henoch-Schönlein, vasculite crioglobulinêmica, vasculite lúpica, induzida por drogas, entre outras.
As vasculites pauci-imunes apresentam como característica principal a inexistência ou escassez de depósitos de imunoglobulinas na parede vascular e em alças capilares glomerulares.10,12
Esse tipo de vasculite está frequentemente associada à existência de ANCA, e, na histopatologia, observa-se uma glomerulonefrite crescêntica necrotizante. Nessa categoria, estão incluídas a GW, a PAM, a síndrome de Churg-Strauss (SCS) e a vasculite pauci-imune limitada ao rim (VLR).
Davies e colaboradores13 observaram, em 1982, oito pacientes com um autoanticorpo denominado ANCA, sugerindo que o ANCA resultava da infecção contra o arbovírus ross river.
Em 1985, van der Woude e colaboradores14 apontaram uma forte associação entre ANCA e GW, estabelecendo o ANCA como um novo biomarcador de algumas formas de vasculite de pequenos vasos, o que foi confirmado posteriormente por Jennette e colaboradores,15 em 1989, em uma coorte de pacientes com vasculite pauci-imune. No contexto das vasculites, o ANCA liga-se a dois antígenos específicos encontrados nos grânulos dos lisossomas de neutrófilos e monócitos, a proteinase-3 (PR-3)16 e a mieloperoxidase (MPO).17
Atualmente, o valor diagnóstico de ensaios para ANCA está bem estabelecido e é bastante aceito não só para GW e PAM, mas também para SCS e VLR.18
Recentemente, sugere-se que a geração do ANCA decorra de uma disfunção na apoptose dos neutrófilos, quando os constituintes dos grânulos translocam-se para a superfície celular e a célula apoptótica é fagocitada por macrófagos e células dendríticas, com ativação de linfócitos T específicos, resultando em autoimunidade. O retardo no clearance dos neutrófilos apoptóticos gera secundariamente necrose e inflamação.19
A patogenicidade dos anticorpos anti-MPO, tanto in vitro20 como in vivo,21 e também dos anticorpos anti-PR-3 in vitro22 e in vivo,23 foram bem observadas. O ANCA ativa os neutrófilos e os monócitos, ocasionando estresse oxidativo e formação de espécies ativas de oxigênio, com liberação de enzimas lisossômicas dos grânulos dos neutrófilos e secreção de citocinas e quimiocinas pró-inflamatórias. Também existem evidências de que o ANCA causa dano às células endoteliais, com adesão e recrutamento de células inflamatórias.22-24
Há mais de 10 anos foi apontado um novo antí-geno-alvo para o ANCA, uma proteína de membrana glicosilada, a lysosomal-associated membrane protein-2 (LAMP-2), existente nos grânulos dos neutrófilos junto com MPO e PR-3. A LAMP-2 é uma proteína que mantém a atividade microbicida dos fagossomos e regula a maturação, sendo responsável pela depuração de agentes infecciosos. Kain e colaboradores,25 em estudo recente, verificaram a existência de LAMP-2 em 14 de 16 pacientes com vasculite necrotizante ativa, sugerindo que LAMP-2 é um outro antígeno-alvo em casos de glomerulonefrite associada a ANCA e um provável iniciador do mecanismo da doença. Os anticorposanti-LAMP-2 apresentam potencial patogênico, pois ativam neutrófilos e causam dano à célula endotelial in vitro e in vivo em ratos.25 O epítopo do LAMP-2 evidencia uma sequência polipeptídica (P41-49) homóloga à de fímbrias de várias bactérias gram-negativas, exceto MPO ou PR-3.26
Por imunofluorescência indireta, utilizando neutrófilos fixados em etanol, pode-se definir três padrões distintos de ANCA:
•c-ANCA (citoplasmático): fluorescência granular citoplasmática difusa. Aproximadamente 90% reagem contra a PR-3 observada nos grânulos de neutrófilos e monócitos.
•p-ANCA (perinuclear): fluorescência perinuclear. Cerca de 90% reagem contra a mieloperoxidase (MPO).
•ANCA atípico: fluorescência inespecífica.
Embora não exista especificidade diagnóstica desses três tipos de ANCA para uma determinada variante de vasculite, a maioria dos pacientes com GW apresenta c-ANCA, enquanto os com PAM e VLR, p-ANCA. Na Tabela 87.1, constam as frequências aproximadas dos dois tipos de ANCA em casos de vasculites pauci-imunes.
O ANCA também tem sido observado em pacientes com diversas doenças, como lúpus eritematoso sistêmico, doenças inflamatórias intestinais, artrite reumatoide, Aids, entre outras. O ANCA pode ser positivo em aproximadamente 13% dos casos de glomerulonefrites mediadas por imunocomplexos.
O valor preditivo positivo (VPP) do ANCA é elevado em pacientes com evidências clínicas de glomerulonefrite crescêntica (perda rápida de função renal, proteinúria, hematúria e anemia) ou outra manifestação de vasculite de pequenos vasos, como púrpura e hemorragia pulmonar.27 Ao contrário, o VPP é baixo nos pacientes sem outras manifestações de vasculite, com função renal normal e somente proteinúria e hematúria.
O ANCA é negativo em pacientes com poliarterite nodosa, arterite de Takayasu e arterite de células gigantes.
662
Na prática, é muito difícil diferenciar clinicamente a GW e a PAM devido à semelhança entre as manifestações clínicas e patológicas, particularmente quanto à doença renal em que ambas as formas apresentam uma glomerulonefrite necrotizante com crescentes.
A diferença fundamental entre as duas condições é a ocorrência de inflamação com granuloma, principalmente no trato respiratório, que ocorre em casos de GW, e não de PAM. Não é comum, entretanto, que haja granuloma em casos de GW, e a confirmação dele tem se tornado menos significativa, pois o tratamento da GW e da PAM é semelhante.
Dessa forma, havendo um quadro clínico compatível, a existência de ANCA confirma uma vasculite pauci-imune e é suficiente para iniciar o tratamento de indução.29 A probabilidade de GW aumenta se houve rc-ANCA, mas, em casos de PAM, a probabilidade dec-ANCA ou p-ANCA é praticamente igual (40 e 50%, respectivamente) (Tab. 87.1).
Granulomatose de Wegener. Essa granulomatose é uma doença sistêmica com vasculite de pequenos vasos e inflamação granulomatosa que envolve o trato respiratório e os rins, como uma glomerulonefrite necrotizante crescêntica. Em vários estudos, a GW não foi considerada como uma doença necessariamente grave e generalizada, podendo, em alguns casos, ser localizada em apenas um ou dois órgãos. Às vezes pode afetar apenas o trato respiratório superior ou inferior.30 A ocorrência de granuloma, e não de vasculite, é que possibilita o estabelecimento do diagnóstico de GW.
Poliangeite microscópica. Embora a nomenclatura “vasculite associada ao ANCA” (VAA) possa ser duvi dosa, principalmente para pacientes com GW ou SCS sem evidência de vasculite, ela continua sendo útil na prática médica. Etretanto, deve-se utilizar “poliangeíte microscópica” (PAM), em vez de poliarterite microscópica, pois, após a descoberta do ANCA, essa doença é definitivamente diferente da poliarterite nodosa (PAN), muitas vezes chamada de PAN macroscópica ou PAN clássica.
Síndrome de Churg-Strauss. Os critérios para o diagnóstico da síndrome de Churg-Strauss foram estabelecidos principalmente a partir de estudos post-morteme são os seguintes: vasculite necrotizante com infiltração por eosinófilos e granulomas extravasculares.31 Contudo, não há necessidade de identificar granuloma se houver coexistência de vasculite sistêmica, envolvendo dois ou mais órgãos extrapulmonares, com asma e eosinofilia maior do que 1,5 x 109/L.32
As principais características e a prevalência das manifestações de pacientes com GW, PAM e SCS podem ser observadas nas Tabelas 87.2 e 87.3.
As VAA, principalmente a GW e a PAM, com frequência afetam os rins, sendo significativos fatores de morbidade e mortalidade.33 Embora a glomerulonefrite rapidamente progressiva (GNRP), nos pacientes ANCA-positivo (por IF ou Elisa), sugira fortemente uma glomerulonefrite associada a ANCA (GAA), as alterações morfológicas renais continuam sendo o padrão-ouro para um diagnóstico acurado.
A biópsia renal deve ser realizada precocemente e, na maiora dos casos, com urgência para que se possa definir a etiologia e a conduta terapêutica. A demora do resultado que a biópsia proporciona ao clínico determina o prognóstico renal do paciente, como, por exemplo, evolução para método de substituição da função renal ou, se a demora for significativa, permanência em programa crônico de diálise.27,28,34
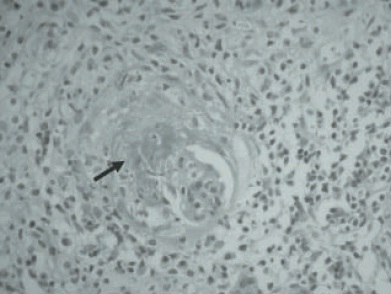
A doença clássica consiste em uma glomerulonefrite necrotizante segmentar e focal com necrose, crescentes celulares e/ou fibrosas e um denso infiltrado inflamatório, conforme ilustrado na Figura 87.1.
Um dado consistente é a relação entre a porcentagem de glomérulos não afetados e o prognóstico. Atualmente, esse achado patológico é considerado o melhor fator preditivo de evolução clínica a curto ou a longo prazo.35
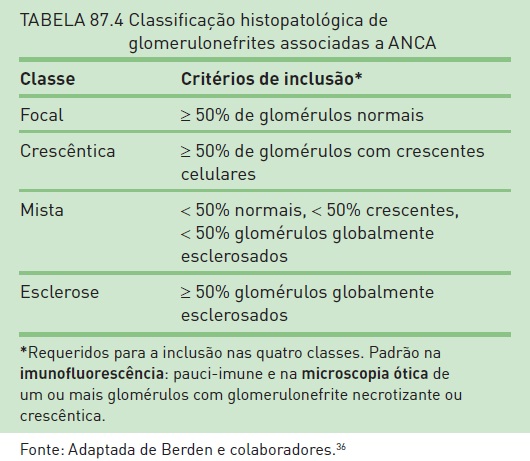
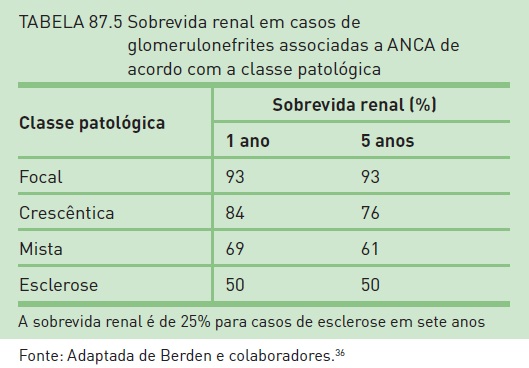
Entretanto, a porcentagem de glomérulos globalmente esclerosados está associada a um prognóstico ruim.34 A porcentagem de crescentes ativas, particularmente as crescentes celulares, está relacionada à recuperação da função renal,28 enquanto a porcentagem de crescentes fibrosas correlaciona-se com um prognóstico ruim.35 Recentemente, Berden e colaboradores36 publicaram uma classificação que possibilita adequada orientação diagnóstica, terapêutica e prognóstica em casos de vasculites com ANCA positivo. Um sumário da classificação patológica e a respectiva sobrevida renal podem ser verificados nas Tabelas 87.4 e 87.5.
Figura 87.1
Glomérulo com necrose central (seta) e uma crescente celular, apresentando denso infiltrado inflamatório no interstício com plasmócitos (cobração de hematoxilina e eosina [HE], magnificação de 200x).
O tratamento de pacientes com vasculites associadas a ANCA é prolongado, devendo haver um equilíbrio na escolha da droga, da dose e do tempo de uso para que se obtenha uma ótima relação risco-benefício.
É fundamental que se identifique com precisão a atividade da vasculite e os pacientes com risco de recidiva. Recidivas podem ocorrer em 30% dos pacientes37 e, em 80% dos casos, nos primeiros 18 meses após a suspensão do tratamento.27 Muitas vezes, a recidiva pode apresentar um quadro clínico igual ao do primeiro episódio, mas, ocasionalmente, pode ocorrer envolvimento de novos órgãos.
Os principais fatores de risco de recidiva são positividade para c-ANCA (PR-3) e envolvimento dos pulmões e das vias aéreas superiores. O risco de recidiva pode aumentar em até quatro vezes se existirem esses três fatores.27
A ideia inicial de que as alterações séricas do ANCA poderiam auxiliar no diagnóstico de atividade da doença não foi confirmada por alguns estudos.38,39 Alguns pacientes em remissão mantêm títulos elevados, enquanto outros apresentam evidência clínica de atividade da vasculite sem elevação dos títulos.
Recomenda-se que, para o diagnóstico de recidiva, sejam realizadas medidas seriadas do ANCA e que os resultados sejam interpretados com os dados clínicos e os demais exames subsidiários.
6664
O sistema de escore de atividade proposto pelo Birmingham Vasculitis Activity Score é eficiente para estabelecer a remissão da doença, mas não informa se a doença permanecerá inativa.40,41
Diversos estudos têm observado que a função renal inicial e a histopatologia são os melhores indicadores da evolução clínica.42,43 De Lind34 considera a porcentagem de glomérulos normais como um forte e possivelmente o melhor preditor histológico da evolução renal a curto e a longo prazo. Inversamente, a alta porcentagem de glomérulos esclerosados está sempre associada a um desenvolvimento desfavorável.
Deve-se suspeitar da recorrência de glomerulonefrite quando houver aumento da hematúria e elevação relativamente rápida da creatinina sérica. Contudo, o aumento da proteinúria, com elevação gradual da creatinina, sem hematúria, indica desenvolvimento de doença crônica, e não sinal de ativação do processo inflamatório. Nesses casos, deve-se realizar biópsia renal e interpretar o resultado junto aos demais dados clínicos a fim de definir o tratamento mais adequado nessa fase da doença.
Alguns estudos foram definitivos para estabelecer a base do conhecimento atual sobre o tratamento de pacientes com VAA.44-50 As VAA, se incorretamente manejadas (p. ex., atraso no tratamento, drogas e doses inadequadas), podem ser fatais e estão associadas a altos índices de morbidade e mortalidade.
O tratamento de pacientes com VAA consiste em três fases: indução, manutenção e tratamento das recidivas.
O principal objetivo, nessa fase, é que o tratamento precoce induza uma rápida remissão da doença com efeitos colaterais mínimos. Para pacientes com quadros graves, o esquema proposto é o seguinte:
Metilprednisolona n A dose desse medicamento é de 7 a 15 mg/kg/dia (máximo de 1 g), por três dias. Após o terceiro dia, deve-se continuar com prednisona, 1 mg/kg/ dia (máximo de 80 mg/dia, no primeiro mês) por quatro a oito semanas (até remissão), com redução gradual da dose. Como opção, após a quarta semana, pode-se utilizar prednisona, em dias alternados, por mais quatro semanas. O período de tempo para a redução da dose deve ser verificado conforme cada paciente, de acordo com a resposta clínica e laboratorial.
Ciclofosfamida n Por muitos anos, a principal discussão sobre essa droga foi a definição do melhor regime: dose oral diária (VO) ou pulsos intravenosos (IV). Em uma metanálise incluindo três estudos randomizados, de Groot e colaboradores43 evidenciaram que a dose cumulativa nos pulsos IV era menor, e essa forma de administração produzia menos efeitos colaterais, particularmente leucopenia e infecções, e facilitava o uso concomitante de mesna para a prevenção de cistite hemorrágica. Apesar de as recidivas manifestarem-se de forma mais frequente com o uso IV, a diferença em relação ao VO não foi estatisticamente significativa, e o estudo original50 não estabeleceu essa diferença.
Atualmente, a melhor evidência sobre a forma de utilizar ciclofosfamida foi publicada em 2009 pelo European Vasculitis Study Group (EUVAS), que recomenda a utilização de ciclofosfamida IV sob a forma de pulsos.50 Os índices de remissão obtidos com a ciclofosfamida IV foram de 88%.
Dose inicial n 0,5 g/m2/superfície corporal (SC) até dose máxima de 1 g/m2/SC. Um hemograma deve ser realizado entre 13 e 15 dias (nadir da leucopenia), objetivando manter o número de leucócitos totais maiores do que 3.000/ mm3. O esquema é repetido a cada 30 dias.
Duração do tratamento n 6 a 12 meses conforme o caso de cada paciente. Devido à toxicidade do uso crônico de ciclofosfamida, a tendência atual é restringir o uso em seis meses, utilizando uma droga oral menos tóxica por mais 12 meses, como azatioprina (ver fase de manutenção).45
Dose n 1,5 a 2 mg/kg/dia. Realiza-se prevenção da leucopenia com hemograma frequente (semanal, após, quinzenal e, a seguir, mensalmente).
Duração do tratamento n 6 a 12 meses conforme o caso de cada paciente. Pode-se utilizar, alternativamente, azatioprina após o sexto mês.
A toxicidade urotelial induzida pela ciclofosfamida, manifestada por cistite hemorrágica, pode ocorrer em 15 a 20% dos pacientes e é mais frequente nos pacientes que administram doses cumulativas elevadas e realizam tratamentos prolongados. Nesses casos, o uso de mesna é recomendado.31
Os demais efeitos colaterais da ciclofosfamida a longo prazo, como insuficiência gonadal (mais frequente em homens), mielossupressão, mielodisplasia e câncer de pele, devem ser monitorados durante e após o tratamento.
Plasmaférese n Existem duas condições que requerem a realização de plasmaférese – hemorragia pulmonar e disfunção renal grave, em pacientes que, na apresentação inicial, dependem de diálise. Um estudo controlado realizado em 48, pacientes com GW, PAM ou VLR observou que a plasmaférese não foi útil para pacientes não dependentes de diálise, mas foi benéfica para os que dependem.51 O estudo do European Vasculitis Study Group (EVAS) publicado na forma de abstract, em 2002, evidenciou que o uso de plasmaférese foi mais efetivo do que o de metilprednisolona em pacientes com insuficiência renal aguda por VAA, apresentando melhora significativa na recuperação da função renal.52 Em 2007, os resultados definitivos foram publicados no estudo MEPEX,47 realizado com 137 pacientes que apresentaram níveis de creatinina maiores do que 5,8 mg/dL, no qual a utilização de plasmaférese, comparada à de metilprednisolona intravenosa, resultou em redução de risco de desenvolvimento de doença renal crônica terminal (DRCT) de 24% em 12 meses (passou de 43 para 19%).
A realização de plasmaférese aumentou o índice de recuperação da função renal, e a sobrevida do paciente e os eventos adversos graves foram semelhantes nos dois grupos. Esses estudos sustentam o uso imediato de plasmaférese em casos de VAA com insuficiência renal aguda grave.
Embora não existam estudos controlados, o uso precoce da plasmaférese está associado à redução de mortalidade nos pacientes com hemorragia pulmonar.53
O protocolo sugerido é iniciar diariamente o procedimento até que não haja mais hemorragia pulmonar e, depois, realizá-lo a cada 48 horas, totalizando 7 a 10 sessões. O esquema ideal seria substituir o plasma com uma solução de albumina a 5% e administrar duas unidades de plasma fresco no final da sessão. A plasmaférese não é indicada para pacientes que não apresentem hemorragia pulmonar.
Rituximabe n A administração de rituximabe está sendo avaliada como tratamento de indução da vasculite ANCA-positivo em dois estudos, o RITUXVAS54 e o RAVE.55 Os resultados iniciais evidenciaram que o uso de rituximabe, comparado ao de ciclofosfamida para indução de remissão da vasculite, apresenta resultados semelhantes. No estudo RITUXVAS, a mortalidade em um ano foi elevada nos dois regimes – 18%. Embora essa droga seja promissora, a evidência ainda é limitada, restringindo-se a pacientes com vasculite refratária à ciclofosfamida. Recomenda-se que esses estudos sejam ampliados e que seja realizada uma análise rigorosa dos desfechos.56,57
666
Sulfametoxazol/trimetropim. O regime terapêutico para os pacientes com doença limitada e sem fatores de prognóstico ruim deve ser constituído por esquemas menos tóxicos. Alguns estudos sugerem o uso de sulfametoxazol/trimetropim em casos de GW como tratamento de indução e eventualmente na manutenção em pacientes com doença limitada. A redução na frequência das infecções induzidas pelo sulfametoxazol/trimetropim, provavelmente pela eliminação do estafilococo áureo, tende a diminuir as recidivas em alguns pacientes.31
Metotrexato. Em estudo não controlado, realizado em pacientes com GW, o uso de metotrexato associado ao de prednisona apresentou índice de remissão comparável aos obtidos com ciclofosfamida,58 mesmo em pacientes com glomerulonefrite,59 mas o índice de recidiva com a administração de metotrexato é elevado – 70% comparado a 47% com ciclofosfamida.60 A maioria dos estudos com essa droga foram efetuados em pacientes com GW que não apresentaram envolvimento renal ou naqueles com glomerulonefrite, mas com função renal preservada.
Corticosteroides. Os corticosteroides foram testados como droga de primeira linha em pacientes sem fatores de prognóstico desfavorável pelo French Vasculitis Study Group.48 A droga induziu remissão prolongada em aproximadamente 50% dos casos, sendo que os demais necessitaram de terapia imunossupressora adicional. Tanto a utilização de azatioprina quanto a de ciclofosfamida foram efetivas em indivíduos resistentes à terapia com corticosteroide. Dessa forma, o uso isolado de corticosteroide não é indicado em casos de VAA devido ao baixo índice de resposta terapêutica.
Micofenolato mofetil. Em um estudo-piloto, a administração de micofenolato mofetil foi segura e eficaz em 9 de 10 pacientes com VAA, mas com doença resistente ou recorrente.61 Em outro estudo recente, realizado em pacientes com GW ou PAM e doença renal leve a moderada, o uso do micofenofato mofetil foi uma alternativa segura tanto no tratamento de indução quanto de manutenção.62 Esses dados, entretanto, ainda são considerados preliminares.
O tratamento de manutenção foi avaliado com várias drogas, incluindo ciclofosfamida, azatioprina, micofenolato mofetil, metotrexato e leflunomida.45,48,49,60 Muitos autores utilizam, nessa fase, a azatioprina, enquanto outros recomendam o uso de metotrexato.
O French Vasculitis Study Group comparou o efeito dessas duas drogas no tratamento de pacientes com GW e PAM, concluindo que nenhuma delas é mais eficiente do que a outra na manutenção da remissão, porém os efeitos colaterais do metotrexato ocorrem com mais frequência e gravidade.48 Um estudo recente do EUVAS, comparando micofenolato mofetil e azatioprina para prevenção de recidivas da VAA, evidenciou que o micofenolato foi menos eficaz nesse desfecho, com mesma proporção de eventos adversos.63
Sendo assim, a tendência é utilizar azatioprina na manutenção, devido à baixa toxicidade e à comprovada eficácia na prevenção de recidiva da VAA.
Não há um consenso na literatura sobre a duração do tratamento com corticosteroide em casos de VAA. O grupo de Chapel Hill,29 na maioria dos casos, suspende a droga após 16 semanas de tratamento, enquanto investigadores europeus utilizam doses baixas por muitos anos.48,49,63
A recidiva da glomerulonefrite crescêntica associada a ANCA ocorre em 30 a 50% dos pacientes que atingem remissão após completar a terapia de indução. A maioria dos pacientes apresenta remissão sustentada ou um ou mais episódios de recidiva, mas, em alguns, a doença persiste com atividade de baixo grau a longo prazo.64 Devido à acentuada toxicidade da imunossupressão prolongada, a escolha da droga e do período de tempo para controle da doença deve ser realizada de forma individual, sendo, muitas vezes, um desafio para o clínico.65
O tratamento de recidivas infrequentes na intensidade de um quadro agudo grave de GNRP consiste em repetir o protocolo inicial, utilizando-se ciclofosfamida IV, metilprednisolona IV, seguido de prednisona VO e plasmaférese, se indicado. Nos pacientes que apresentam alto índice de recidivas, recidiva precoce ou que mantêm alguma atividade de doença clínica e laboratorial, indica-se tratamento de manutenção. A administração de azatioprina é uma opção segura e menos tóxica, devendo-se considerar o uso de micofenolato mofetil e rituximabe como alternativas para a doença persistente ou refratária.65
Análises a longo prazo de pacientes com VAA evidenciaram que idade superior a 50 anos e envolvimento pulmonar e renal aumentam quatro vezes o risco de doença renal crônica em estágio terminal (DRCT).
Em relação à sobrevida do paciente, a hemorragia pulmonar aumenta nove vezes o risco relativo de morte e quatro vezes quando os ANCA são anti-PR3. No estudo de Hogan e colaboradores,66 nível de creatinina sérica inicial, afrodescendência e esclerose arterial na biópsia foram fortes preditores de menor sobrevida renal.
Entretanto, 50% dos pacientes que dependem de diálise restabelecem a função renal com o tratamento, o que justifica tratar todos os pacientes independentemente do nível de creatinina inicial. O risco de desenvolvimento de DRCT é exatamente a mudança na taxa de filtração glomerular nos primeiros quatro meses de tratamento.64 Outro fator importante é a resistência ao tratamento imunossupressor, que foi preditiva de progressão para DRCT em média dois meses após o início da terapia de indução.65
O quadro clínico dessa paciente do sexo feminino, com mais de 50 anos de idade, sintomas e sinais sistêmicos (febrícula, fadiga, emagrecimento), episódios de hemoptise, oligoanúria e congestão pulmonar sugere uma vasculite sistêmica manifestada pela síndrome pulmão-rim. Os exames laboratoriais confirmam o diagnóstico de glomerulonefrite rapidamente progressiva (insuficiência renal grave, hematúria, proteinúria, cilindros hemáti- cos), e o raio X de tórax com infiltrado alveolointersticial bilteral indica alveolite hemorrágica, possivelmente associada à congestão pulmonar pela insuficiência renal oligoanúrica. A ocorrência de anemia acentuada é comum na fase aguda da doença. Indica-se tratamento imediato da insuficiência renal aguda com hemodiálise, e, nesse momento, é importante a indicação precoce de biópsia renal para definir o diagnóstico histológico. A positividade para c-ANCA em título alto e a histologia evidenciando uma glomerulonefrite necrotizante segmentar com crescentes caracterizam uma vasculite sistêmica de pequenos vasos ANCA-positivo (VAA) em atividade. É incomum a existência de granuloma no tecido renal ou respiratório, que confirmaria o diagnóstico etiológico de granulomatose de Wegener, hipótese mais provável pelo gênero, pela faixa etária da paciente e pelo tipo de ANCA (citoplasmático). O tratamento específico em casos de VAA, na fase de indução, deve ser imediato e consiste em pulsoterapia com metilprednisolona, 1 g, IV, por três dias, seguido de prednisona oral, 1 mg/ kg/dia, durante seis semanas, com redução progressiva até 5 mg/dia, e plasmaférese (7 a 10 sessões), em seguida, ciclofosfamida sob a forma de pulsoterapia, na dose inicial de 0,5 g/m2 SC (filtração glomerular menor do que 30 mL/min) e, após, doses mensais de 0,5 a 1 g/m2 SC, até o sexto mês. A avaliação dos seios da face por tomografia computadorizada identifica sinusite aguda, tratada com sulfametoxazol associado a trimetropim, que são mantidos profilaticamente a fim de reduzir a recidiva da VAA. Após três semanas do início do tratamento, a paciente apresenta melhora progressiva da função renal, e a diálise é suspensa. A função renal é restabelecida no segundo mês, e, no sexto mês, é iniciado o tratamento de manutenção com azatioprina em substituição à ciclofosfamida. O uso de prednisona é suspenso no sexto mês. Após 18 meses, a administração de azatioprina é suspensa, e a paciente mantém creatinina de 1,1 mg/dL. É importante monitorar sinais clínicos e laboratoriais de recidiva da VAA nessa paciente; caso ela ocorra na mesma intensidade que o surto inicial, deve ser tratada com protocolo semelhante.
1.Hoffman GS, Kerr GS, Leavitt RY, Hallahan CW, Lebovics RS, Travis WD, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med. 1992;116(6):488-98.
2.Watts RA, Scott DG. Epidemiology of the vasculitides. Curr Opin Rheumatol. 2003;15(1):11-6.
3.Reinhold-Keller E, Herlyn K, Wagner-Bastmeyer R, Gutfleisch J, Peter HH, Raspe HH, et al. No difference in the incidence of vasculitides between north and south Germany: First results of the German vasculites register. Rheumatology (Oxford). 2002;41(5):540-9.
4 Jennette JC, Falk RJ, Andrassy K, Bacon PA, Churg J, Gross WL, et al. Nomenclature of systemic vasculitides. Proposal of an International Consensus Conference. Arthritis Rheum. 1994;37(2):187-92.
5.Lie JT. Classification of systemic vasculitis. In: Churg A, Churg J, editors. Systemic vasculitides. New York: Igaku-Shoin; 1991. p. 159-79.
6.Churg J. Large vessel vasculites. Clin Exp Immunol. 1993;93 Suppl1:11-2.
7.Kirkland GS, Savige J, Wilson D, Heale W, Sinclair RA, Hope RN. Classical polyarteritis nodosa and microscopic polyarteritis with medium vessel involvement: a comparison of the clinical and laboratory features. Clin Nephrol. 1997;47(3):176-80.
8.Gribetz D, Landing B, Larson E. Kawasaki disease: mucocutaneous lymph node syndrome (MCLNS). In: Churg A, Churg J, editors. Systemic vasculitides. New York: Igaku-Shoin; 1991. p. 257-72.
9.Jennette JC, Falk RJ. The pathology of vasculitis involving the kidney. Am J Kidney Dis. 1994;24(1):130-41.
10.Jennette JC, Falk RJ. Small-vessel vasculitis. N Engl J Med.1997;337(21):1512-23.
11.Jennette JC, Tomas BD, Falk RJ. Microscopic polyangiitis (microscopic polyarteritis). Semin Diagn Pathol. 2001;18(1):3-13.
12.Ronco P, Mougenot B, Bindi P, Noel LH, Mignon F, Lesavre P. Idiopathic extracapillary glomerulonephritides without immune deposits are vascularitides: clinical and serologic analysis. Bull Acad Natl Med.1993;177(3):481-94.
13.Davies DJ, Moran JE, Niall JF, Ryan GB. Segmental necrotizing glomerulonephritis with antineutrophil antibody: possible arbovirus aetiology? Br Med J (Clin Res Ed). 1982;285(6342):606.
14.van der Woude FJ, Rasmussen N, Lobatto S, Wiik A, Permin H, van Es LA, et al. Autoantibodies against neutrophils and monocytes: tool for diagnosis and marker of disease activity in Wegener’s granulomatosis. Lancet. 1985;1(8426):425-9.
15.Jennete JC, Wilkman AS, Falk RJ. Anti-neutrophil cytoplasmicautoantibody-associated glomerulonephritis and vasculitis. Am J Pathol.1989;135(5):921-30.
16.Goldschmeding R, van der Schoot CE, ten Bokkel Huinink D, Hack CE, van den Ende ME, Kallenberg CG, et al. Wegener’s granulomatosis autoantibodies identify a novel diisopropylfluorophosphate-binding protein in the lysosomes of normal human neutrophils. J Clin Invest.1989;84(5):1577-87.
17.Falk RJ, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies with specificity for myeloperoxidase in patients with systemic vasculitis and idiopathic necrotizing and crescentic glomerulonephritis. N Engl J Med.1988;318(25):1651-7.
18.Hagen EC, Daha MR, Hermans J, Andrassy K, Csernok E, Gaskin G, et al. Diagnostic value of standardized assays for anti-neutrophil cytoplasmic antibodies in idiopathic systemic vasculitis. EC/BCR Project for ANCA Assay Standardization. Kidney Int. 1998;53(3):743-53.
19.Yang JJ, Tuttle RH, Hogan SL, Taylor JG, Phillips BD, Falk RJ, et al. Target antigens for anti-neutrophil cytoplasmic auto-antibodies (ANCA) are on the surface of primed and apoptotic but not unstimulated neutrophils. Clin Exp Immunol. 2000;121(1):165-72.
20.Xiao H, Heeringa P, Hu P, Liu Z, Zhao M, Aratani Y, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest.2002;110(7):955-63.
21.Little MA, Smyth CL, Yadav R, Ambrose L, Cook HT, Nourshargh S, et al. Antineutrophil cytoplasmic antibodies directed against myeloperoxidase augment leukocyte-microvascular interactions in vivo. Blood.2005;106(6):2050-8.
22.Falk RJ, Terrell RS, Charles LA, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci U S A. 1990;87(11):4115-9.
23.Pfister H, Ollert M, Fröhlich LF, Quintanilla-Martinez L, Colby TV, Specks U, et al. Antineutrophil cytoplasmic autoantibodies against the murine homolog of proteinase 3 (Wegener autoantigen) are pathogenic in vivo. Blood. 2004;104(5):1411-8.
24.Gomez-Puerta JA, Bosch X. Anti-neutrophil cytoplasmic antibody pathogenesis in small vessel vasculitis: an update. Am J Pathol.2009;175(5):1790-8.
25.Kain R, Exner M, Brandes R, Ziebermayr R, Cunningham D, Alderson CA, et al. Molecular mimicry in pauci-immune focal necrotizing glomerulonephritis. Nat Med. 2008;14(10):1088-96.
26.Salama AD, Pusey CD. Shining a LAMP on pauci-immune focal segmental glomerulonephritis. Kidney Int. 2009;76(1):15-7.
27.Hauer HA, Bajema IM, Van Houwelingen HC, Ferrario F, Noël LH, Waldherr R, et al. Determinants outcome in ANCA-associated glomerulonephritis: a prospective clinico-histopatological analysis of 96 patients. Kidney Int. 2002;62(5):1732-42.
28.Nachman PH, Jennette JC, Falk RJ. Vasculitic disease of the kidney. In: Schrier RW, editor. Diseases of the kidney and urinary tract. 7th ed. Philadelphia: Lippincott Williams & Wilkins; 2007. p. 1748-75.
29.Falk RJ, Jennette JC. ANCA disease: where is this field heading? J Am Soc Nephrol. 2010;21(5):745-52.
30.Stegeman CA, Kallenberg CMK. Pathogenesis of angiitis. In: Davison AM, Cameron JS, Grunfeld JP, Ponticelli C, Ritz E, Christopher G, et al., editors. Oxford textbook of clinical nephrology. 3rd ed. New York: Oxford University; 2005. p. 741-52.
31.Gaskin G. Systemic Vasculitis. In: Davison AM, Cameron JS, Grunfeld JP, Ponticelli C, Ritz E, Christopher G, et al., editors. Oxford textbook of clinical nephrology. 3rd ed. New York: Oxford University; 2005. p. 766-96.
32.Lanham JG, Elkon KB, Pusey CD, Hughes GR. Systemic vasculitis with asthma and eosinophilia: a clinical approach to the Churg-Strausssyndrome. Medicine (Baltimore). 1984;63(2):65-81.
33.Mukhtyar C, Flossmann O, Hellmich B, Bacon P, Cid M, Cohen-Tervaert JW, et al. Outcomes from studies of antineutrophil cytoplasm antibody associated vasculitis. A systematic review by the European League Against Rheumatism systemic vasculitis task force. Ann Rheum Dis.2008;67(7):1004-10.
34.de Lind van Wijngaarden RA, Hauer HA, Wolterbeek R, Jayne DR, Gaskin G, Rasmussen N, et al. Clinical and histologic determinants of renal outcome in ANCA-associated vasculitis. A prospective analysis of 100 patients with severe renal involvement. J Am Soc Nephrol.2006;17(8):2264-74.
35.de Lind van Wijngaarden RA. ANCA-associated glomerulonephritis: insights into etiology, pathogenesis and prognosis [tese]. Leiden: Leiden University Medical Center; 2009. p. 88-103.
36.Berden AE, Ferrario F, Hagen EC, Jayne DR, Jennette JC, Joh K, et al. Histopathologic classification of ANCA-associated glomerulonephritis. J Am Soc Nephrol. 2010;21(10):1628-36.
37.Nachman PH, Hogan SL, Jennette JC, Falk RJ. Treatment response and relapse in antineutrophil cytoplasmic autoantibody-associated microscopic polyangiitis and glomerulonephritis. J Am Soc Nephrol. 1996;7(1):33-9.
38.Boomsma MM, Stegeman CA, van der Leij MJ, Oost W, Hermans J, Kallenberg CG, et al. Prediction of relapses in Wegener’s granulomatosis by measurement of antineutrophil cytoplasmic antibody levels: a prospective study. Arthritis Rheum. 2000;43(9):2025-33.
39.Ara J, Mirapeix E, Rodriguez R, Saurina A, Darnell A. Relationship between ANCA and disease activity in small vessel vasculitis patients withanti-MPO ANCA. Nephrol Dial Transplant. 1999;14(7):1667-72.
40.Luqmani RA, Bacon PA, Moots RJ, Janssen BA, Pall A, Emery P, et al. Birmingham Vasculitis Activity Score (BVAS) in systemic necrotizing vasculitis. QJM. 1994;87(11):671-8.
41.Stone JH, Hoffman GS, Merkel PA, Min YI, Uhlfelder ML, Hellmann DB, et al. A disease-specific activity index for Wegener’s granulomatosis: modification of the Birmingham Vasculitis Activity Score. International Network for the Study of the Systemic Vasculitides (INSSYS). Arthritis Rheum. 2001;44(4):912-20.
42.Neumann I, Kain R, Regele H, Soleiman A, Kandutsch S, Meisl FT. Histological and clinical predictors of early and late outcome in ANCA-associated vasculitis. Nephrol Dial Transplant. 2005;20(1):96-104.
43.de Groot K, Adu D, Savage CO; EUVAS (European vasculitis study group). The value of pulse cyclophosphamide in ANCA associated vasculitis: meta-analysis and critical review. Nephrol Dial Transplant.2001;16(10):2018-27.
44.Falk RJ, Hogan S, Carey TS, Jennette JC. Clinical course of anti-neutrophil cytoplasmic autoantibody-associated glomerulonephritis and systemic vasculitis. The Glomerular Disease Collaborative Network. Ann Intern Med. 1990;113(9):656-63.
45.Jayne D, Rasmussen N, Andrassy K, Bacon P, Tervaert JW, Dadoniené J, et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med.2003;349(1):36-44.
46.De Groot K, Rasmussen N, Bacon PA, Tervaert JW, Feighery C, Grego- rini G, et al. Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody-associatedvasculitis. Arthritis Rheum. 2005;52(8):2461-9.
47.Jayne DR, Gaskin G, Rasmussen N, Abramowicz D, Ferrario F, Guillevin L, et al. Randomized trial of plasma exchange or high dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol. 2007;18(7):2180-8.
48.Pagnoux C, Mahr A, Hamidou MA, Boffa JJ, Ruivard M, Ducroix JP, et al. Azathioprine or methotrexate maintenance for ANCA-associatedvasculitis. N Engl J Med. 2008;359(26):2790-803.
49.Hiemstra T, Walsh M, de Groot K, Huser T, Mahr A, Pagnoux C, et al. Randomized trial of mycophenolate mofetil vs. azathioprine for maintenance therapy in ANCA-associated vasculitis (IMPROVE). APMIS. 2009;117(Suppl. 127):77-8.
50.de Groot K, Harper L, Jayne DR, Flores Suarez LF, Gregorini G, Gross WL, et al. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med. 2009;150(10):670-80.
51.Pusey CD, Rees AJ, Evans DJ, Peters DK, Lockwood CM. Plasma exchange in focal necrotizing glomerulonephritis without anti-GBMantibodies. Kidney Int. 1991;40(4):757-63.
52.Gaskin G, Jayne DR. European Vasculitis Study Group: adjunctive plasma exchange is superior to methylprednisolone in acute renal fai- lure due to ANCA-associated glomerulonephritis. J Am Soc Nephrol. 2002;13:2A.
53.Klemmer PJ, Chalermskulrat W, Reif MS, Hogan SL, Henke DC, Falk RJ. Plasmapheresis therapy for diffuse alveolar hemorrhage in patients withsmall-vessel vasculitis. Am J Kidney Dis. 2003;42(6):1149-53.
54.Jones R, Walsh M, Jayne D. European Vasculitis Study Group: randomised trial of rituximab versus cyclophosphamide for ANCA-associatedrenal vasculitis: RITUXVAS. J Am Soc Nephrol. 2008;19:61A.
55.Stone JH, Merkel PA, Seo P. The RAVE-ITN Research Group: Rituximab versus cyclophosphamide for induction of remission in ANCA-associated vasculitis: a randomized controlled trial (RAVE). Arthritis Rheum. 2009;60(Suppl):S204.
56.Jones RB, Tervaert JW, Hauser T, Luqmani R, Morgan MD, Peh CA, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med. 2010;363(3):211-20.
57.Stone JH, Merkel PA, Spiera R, Seo P, Langford CA, Hoffman GS, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363(3):221-32.
58.Sneller MC, Hoffman GS, Talar-Williams C, Kerr GS, Hallahan CW, Fauci AS. An analysis of forty-two Wegener’s granulomatosis patients treated with methotrexate and prednisone. Arthritis Rheum.1995;38(5):608-13.
59.Langford CA, Talar W, Sneller MC. Use of methotrexate and glu- cocorticoids in the treatment of Wegener’s granulomatosis. Long-term renal outcome in patients with glomerulonephritis. Arthritis Rheum.2000;43(8):1836-40.
60.Metzler C, Miehle N, Manger K, Iking-Konert C, de Groot K, Hell- mich B, et al. Elevated relapse rate under oral methotrexate versus leflunomide for maintenance of remission in Wegener’s granulomatosis. Rheumatology (Oxford). 2007;46(7):1087-91.
61.Joy MS, Hogan SL, Jennette JC, Falk RJ, Nachman PH. A pilot study using mycophenolate mofetil in relapsing or resistant ANCA small vessel vasculitis. Nephrol Dial Transplant. 2005;20(12):2725-32.
62.Silva F, Specks U, Kalra S, Hogan MC, Leung N, Sethi S, et al. Mycophenolate mofetil for induction and maintenance of remission in microscopic polyangiitis with mild to moderate renal involvement: a prospective, open-label pilot trial. Clin J Am Soc Nephrol. 2010;5(3):445-53.
63.Hiemstra TF, Walsh M, Mahr A, Savage CO, de Groot K, Harper L, et al. Mycophenolate vs. azathioprine for remission maintenance in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized controlled trial. European Vasculitis Study Group (EUVAS). JAMA.2010;304(21):2381-8.
64.Hogan SL, Falk RJ, Chin H, Cai J, Jennette CE, Jennette JC, et al. Predictors of relapse and treatment resistance in anti-neutrophil cytoplasmic antibody-associated small-vessel vasculitis. Ann Inter Med.2005;143(9):621-31.
65.Lionaki S, Jennette JC, Falk RJ. Anti-neutrophil cytoplasmic (ANCA) and anti-glomerular basement membrane (GBM) autoantibodies in necrotizing and crescentic glomerulonephritis. Semin Immunopathol.2007;29(4):459-74.
66.Hogan SL, Nachman PH, Wilkman AS, Jennette JC, Falk RJ. Prognostic markers in patients with anti-neutrophil cytoplasmic auto-antibody-associated microscopic poliangiitis and glomerulonephritis. J Am Soc Nephrol. 1996;7(1):23-32.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.