(Carregando Índice)... (Carregando Índice)... |
Autores:
Ane Cláudia Fernandes Nunes
Bióloga geneticista. Pesquisadora do Laboratório de Nefrologia Celular, Genética e Molecular (LIM-29) da Faculdade de Medicina da Universidade de São Paulo (USP). Mestre e Doutora em Ciências Médicas: Nefrologia pela UFRGS. Pós-doutora pelo Instituto de Biofísica Carlos Chagas Filho da UFRJ. Pós-doutora pelo Departamento de Clínica Médica da Faculdade de Medicina da USP.
Elvino Barros
Médico nefrologista. Professor associado da Faculdade de Medicina da UFRGS. Doutor em
Nefrologia pela Universidade Federal de São Paulo (UNIFESP).
Última revisão: 25/08/2014
Comentários de assinantes: 0
Um paciente do sexo masculino, 24 anos, branco, após acidente de trânsito com motocicleta, foi atendido no hospital, apresentando múltiplas escoriações. A partir da avaliação clínica e radiológica, foi verificado que não havia fraturas ou outras lesões significativas, e o paciente teve alta da emergência. Antes de deixar a sala da emergência, o paciente observou que sua urina estava avermelhada.
O médico plantonista foi comunicado e suspeitou que houvesse lesão renal traumática; por isso, encaminhou o paciente para realização de arteriografia de urgência. O exame evidenciou apenas múltiplos cistos renais. O paciente recebeu alta da emergência com orientação de continuar o acompanhamento nefrológico. Foi internado, no dia seguinte, em outro hospital para investigação, e obteve diagnóstico de contusão abdominal. Apresentou dores generalizadas e dor lombar a direita.
Na urografia excretora e na cistografia, foi observado quadro radiológico de rins policísticos, sem evidência de outras alterações significativas nos rins, nos ureteres e na bexiga. Na historia familiar, relatou que sua mãe era portadora de nefropatia e necessitava de hemodialise, tendo falecido de carcinoma de mama aos 67 anos de idade. A mãe era portadora de hipertensão arterial sistêmica desde os 25 anos de idade e iniciou programa regular de hemodialise aos 44 anos.
A doença renal policística autossômica dominante (DRPAD) consiste na nefropatia hereditária mais comum, sendo caracterizada principalmente por desenvolvimento e crescimento bilateral de cistos renais globulares e focais que aumentam em número e tamanho conforme a idade.
Embora o comprometimento renal seja a característica mais acentuada, há um número variado de desfechos sistêmicos potenciais associados à DRPAD que devem ser considerados na investigação diagnóstica. O envolvimento hepático, pancreático, cerebrovascular e/ou outros sinais e sintomas extrarrenais que possam causar complicações significativas devem ser analisados de forma cuidadosa nos pacientes potencialmente portadores de DRPAD.
Recentemente, os critérios de diagnóstico da DRPAD foram redefinidos por Pei e colaboradores,1 e a adoção de critérios mais precisos referentes às faixas etárias dos pacientes aumentou o valor preditivo do diagnóstico por exame de imagem por meio da ultrassonografia (Fig. 88.1). Além disso, a inclusão de testes genéticos, como os estudos de ligação gênica ou o sequenciamento direto do DNA, também possibilitaram a identificação de novos casos com mais exatidão. Esses avanços, por sua vez, estão contribuindo de forma crucial para o entendimento da DRPAD, inclusive entre os pacientes que não apresentam história familiar da doença, que totalizam cerca de 10% de todos os casos, sugerindo que a existência de novas mutações nos genes associados à doença é um fator que deve ser considerado no processamento do diagnóstico.

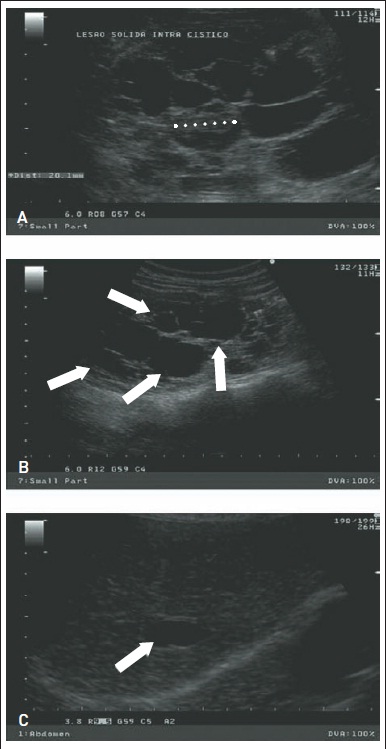
Figura 88.1
Exame de imagem obtido por ultrassonografia evidenciando diversos cistos renais bilaterais e cisto hepático isolado. (A) Múltiplos cistos no rim direito de um paciente adulto do sexo masculino. Nota-se lesão sólida intracística (linha pontilhada) (B) Múltiplos cistos no rim esquerdo do mesmo paciente adulto do sexo masculino. As setas indicam cistos de dimensões variadas. (C) Cisto hepático isolado (seta) observado no mesmo paciente de A e B.
A DRPAD apresenta uma prevalência mundial de 1:400 a 1:1.000, sendo considerada uma das doenças humanas hereditárias mais comuns e a doença renal monogênica mais frequente. Talvez, por essa razão, a DRPAD seja bastante estudada, tanto em termos clínicos quanto em
relação a sua patogênese celular e molecular.
A doença causa cerca de 3 mil hospitalizações por ano, nos Estados Unidos, determinando a realização de diálise crônica em 8 a 10% da população norte-americana. Na Europa, entre 9 e 10% dos pacientes submetidos à hemodiálise apresentam rins policísticos. Na Ásia, por sua vez, é interessante observar que a prevalência é menor, sendo diagnosticada essa condição somente em 2,5 a 3,2% dos pacientes em tratamento dialítico. Aproximadamente 5.000 a 6.000 novos casos da doença são diagnosticados por ano, nos Estados Unidos, sendo 40% desses casos descobertos em torno dos 45 anos.
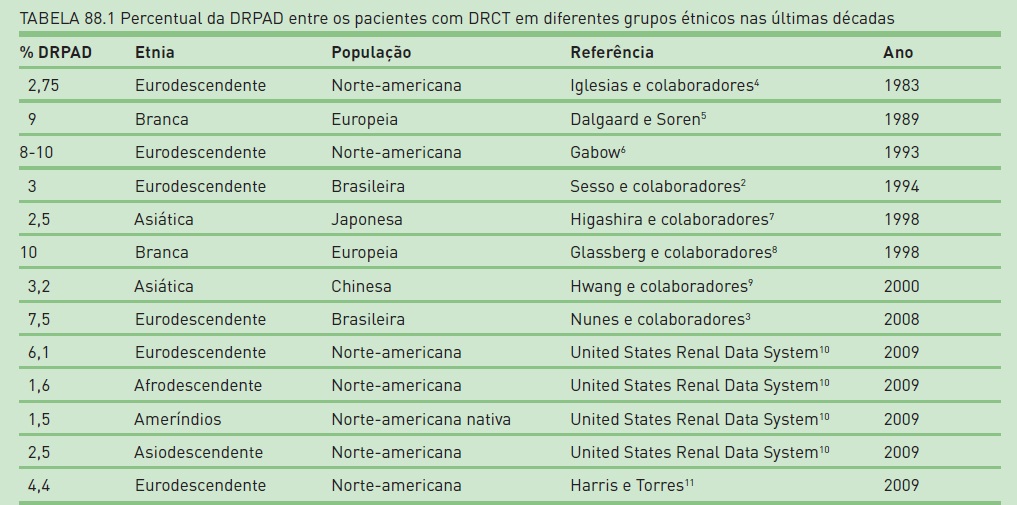
No Brasil, estima-se a existência de cerca de 60 mil pacientes em tratamento para doença renal crônica terminal (DRCT). De acordo com o levantamento realizado, em 1991, na grande São Paulo, em um total de 2.905 pacientes analisados, 87 apresentaram rins policísticos como diagnóstico, submetendo 3% do total de pacientes a tratamento dialítico.2 Outro estudo brasileiro que foi desenvolvido no sul do Brasil, efetuado com pesquisa a campo, aponta que 7,5% dos pacientes em programa dialítico são afetados pela DRPAD.3 No entanto, esse dado difere do obtido em estudo realizado na população norte-americana no mesmo período, e o United States Renal Data Service atribui 3% para o valor aproximado dos casos de DRPAD identificados entre os pacientes com DRCT no ano de 2009. Ressalta-se que o índice norte-americano foi obtido a partir de análises de banco de dados públicos, e não em uma pesquisa detalhada a campo. Desse forma, deve-se considerar que o número de casos de DRPAD, na maioria das análises apresentadas, é subestimado.
De um modo geral, são necessários dados relativos às populações africanas ou afrodescendentes, e também são escassos os dados sobre populações latino-americanas.
Alguns dados epidemiológicos da doença renal policística constam na Tabela 88.1.
Aproximadamente 50% dos pacientes com DRPAD desenvolvem DRCT até os 60 anos. Na maioria dos casos, o acompanhamento clínico regular é fundamental, e a adoção de um método de terapia renal substitutiva (TRS – hemodiálise, diálise peritoneal ou transplante) torna-se indispensável no tratamento.
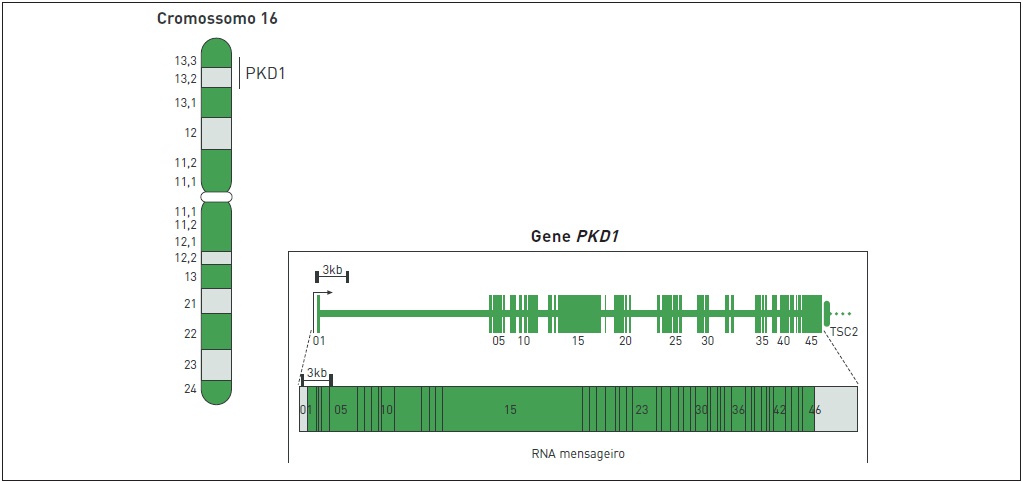
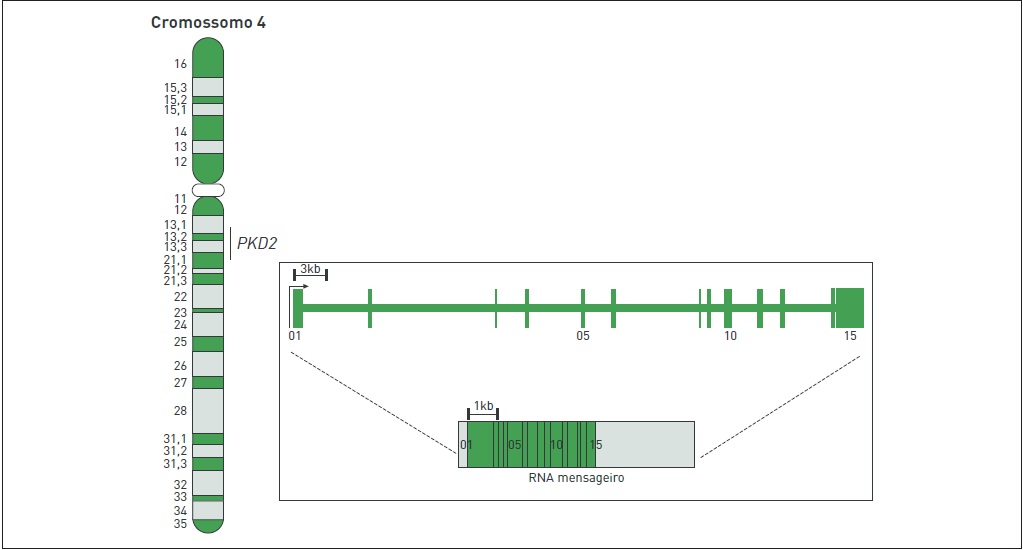
Embora apresente uma penetrância presumível de 100%, a DRPAD é considerada heterogênea geneticamente, sendo causada por mutações em um dos dois genes sabidamente associados à doença: polycystic kidney disease 1 (PKD1), localizado na região cromossômica 16p13.3, e polycystic kidney disease 2 (PKD2), mapeado em 4q21. As Figuras 88.2 e 88.3 ilustram esses genes. A maioria dos casos (80 a 85%) resulta de mutações em PKD1, causando a DRPAD tipo 1 (DRPAD1). Nos demais pacientes (15 a 20%), as mutações são identificadas em PKD2 e originam
a DRPAD tipo 2 (DRPAD2).
A existência de um lócus adicional relacionado à DRPAD foi sugerida por estudos realizados em famílias relatadas como não ligadas exclusivamente a PKD1 ou a PKD2. Contudo, deve-se atentar para essa hipótese, pois os casos que a sustentavam parecem, na verdade, representar condições peculiares da expressão dos genes PKD1 e PKD2, como a identificação de heterozigotos compostos para mutações em ambos os genes PKD1 e PKD2. Além disso, faltam confirmações dos achados observados para as demais famílias referidas como não ligadas a esses lócus.
Figura 88.2
Representação esquemática da localização cromossômica e da estrutura do gene PKD1.
Figura 88.3
Representação esquemática da localização cromossômica e da estrutura do gene PKD2.
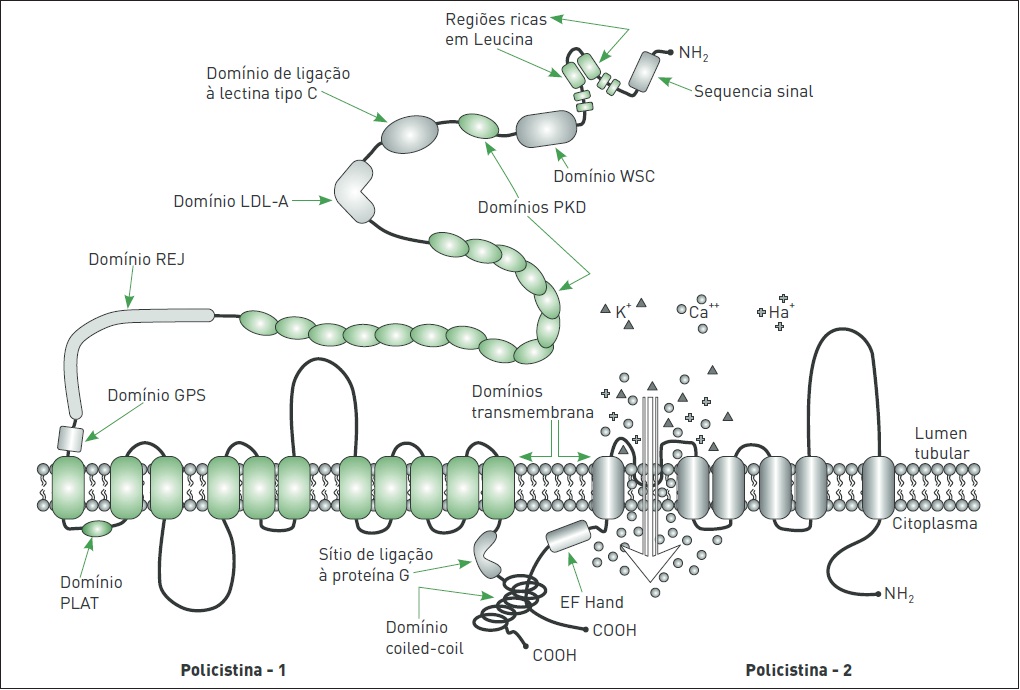
O gene PKD1 abrange 46 éxons distribuídos ao longo de um segmento genômico com cerca de 52 kb (Fig. 88.2), produzindo um RNA mensageiro de 14,2 kb e associado a um quadro de leitura aberta de aproximadamente 12,9 kb. O gene PKD1 codifica a policistina-1 (PC1), uma glicoproteína integral de membrana de 4.303 aminoácidos.
A PC1 apresenta uma grande porção extracelular aminoterminal, com cerca de 3.000 aminoácidos, 11 domínios transmembrana e uma porção carboxiterminal intracelular curta. A porção extracelular apresenta uma combinação complexa de domínios, aparentemente envolvidos em interações proteína-proteína e proteína-carboidrato.
Esse conjunto de domínios compreende 16 cópias de uma repetição de 80 aminoácidos semelhante a regiões da imunoglobulina, os domínios PKD, uma sequência sinal, segmentos de repetições ricas em leucina, um domínio WSC, domínios de ligação à lectina tipo C e LDL-A, além de um domínio receptor for egg jelly (REJ) e um domínio G-protein-coupled receptor proteolytic site (GPS).
Alguns domínios apresentam características particularmente importantes. O domínio REJ, por exemplo, parece desempenhar um papel regulatório na molécula, enquanto o domínio GPS parece ser essencial para a clivagem da proteína, um processo com consequências estruturais e funcionais renais significativas. A porção carboxiterminal da PC1, por sua vez, contém diversos sítios de fosforilação e um domínio helicoidal envolvido em interações proteína-proteína, denominado coiled-coil.
Esse domínio é responsável pela interação física entre as porções carboxiterminais da policistina-1 e da policistina-2 (PC2), o produto do gene PKD2. Estudos recentes evidenciam que a extremidade carboxiterminal também pode ser clivada e migrar para o núcleo da célula. A existência desses vários componentes estruturais/funcionais indica que a PC1 constitui-se em uma molécula grande e multifuncional, apresentando características tanto de receptor de membrana quanto de molécula de adesão que interage com a PC2 no epitélio tubular.
O gene PKD2 (Fig. 88.3) expressa um RNA mensageiro de 5,4 kb que codifica um polipeptídeo de 968 aminoácidos, a PC2. Esta contém seis domínios transmembrana, e ambas as caudas, amino e carboxiterminais, são intracitoplasmáticas.
Além disso, a PC2 apresenta homologia com os seis últimos domínios transmembrânicos da PC1. Juntas, as policistinas formam uma subfamília à parte (TRPP) dos canais TRP (receptor de potencial transiente). A PC2 funciona, ainda, como um canal de cátions não seletivo permeável a Ca++, cuja atividade é regulada pela PC1.
Destaca-se também a presença de um domínio EF hand na porção carboxiterminal da PC2, envolvido na ligação ao Ca++ (Fig. 88.4).
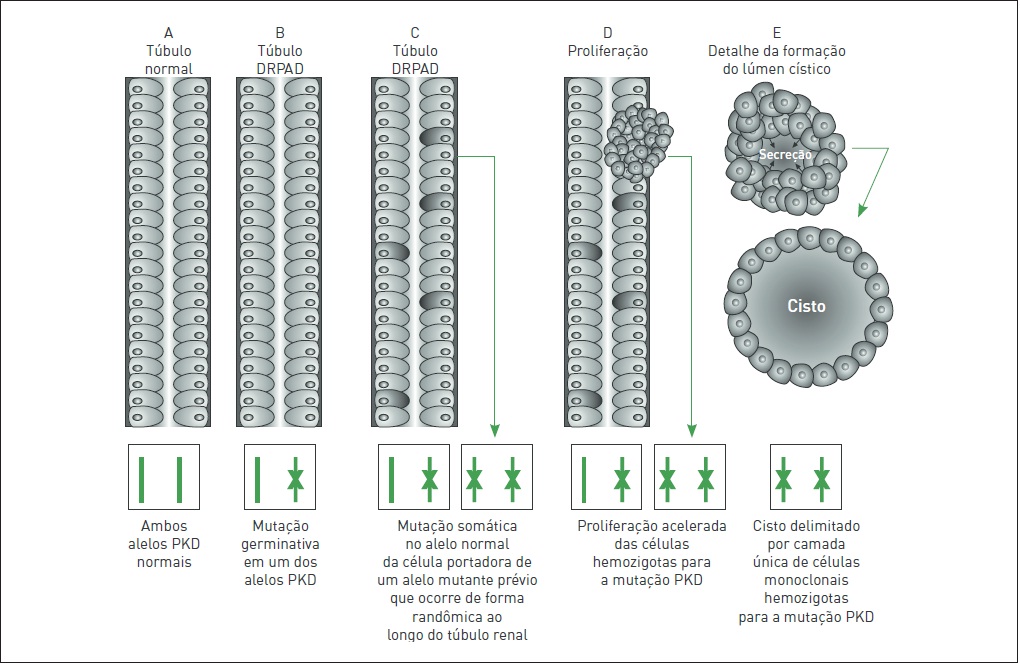
Embora a DRPAD apresente um padrão de herança gênica evidentemente dominante, no âmbito celular/molecular, a expressão dos alelos mutantes envolvidos com a doença é recessiva. Nesse sentido, a hipótese mais aceita propõe que sejam necessários dois eventos mutacionais independentes para o desenvolvimento da DRPAD.
Embora ambas as formas sejam similares em seus fenótipos, a DRPAD1 é mais grave do que a DRPAD2, sendo que as medianas de idade de diagnóstico e de desenvolvimento de DRCT são mais baixas. Além disso, o paciente com DRPAD1 apresenta mais propensão a hipertensão arterial, hematúria e infecções do trato urinário. O desenvolvimento precoce de um número maior de cistos nos pacientes portadores de DRPAD1 parece explicar o fato de essa forma ser mais grave do que a DRPAD2, uma vez que parece não haver diferença entre as duas condições quanto à rapidez de formação e/ou expansão cística. Em termos fenotípicos, a DRPAD apresenta grande variabilidade entre diferentes famílias e também entre membros de uma mesma família. Casos de recém-nascidos de famílias portadoras de DRPAD, com sinais estabelecidos da doença, são bons exemplos dessa variabilidade.
Vários estudos concordam que esse modelo de dois eventos, também conhecido como segundo golpe (second hit), pode explicar o mecanismo focal e heterogêneo da formação cística da DRPAD no rim e no fígado (Fig. 88.5). Esse processo, que pode ser aplicado em ambas as formas genéticas da doença (DRPAD1 e DRPAD2), tem como primeiro golpe a mutação germinativa herdada de um dos progenitores e presente em todas as células tubulares renais do paciente, enquanto o segundo evento é representado por uma mutação somática no alelo previamente normal do gene.
Além do lócus gênico envolvido na doença, a posição intragênica da mutação germinativa e a natureza de algumas mutações podem explicar a variação clínica interfamiliar, mas não o fato de indivíduos com uma mutação germinativa comum apresentarem fenótipos significativamente distintos. O tipo específico da mutação presente parece não se correlacionar com o fenótipo de uma forma determinante, mas mutações localizadas na porção 5’ do gene PKD1 foram associadas à evolução para DRCT mais precoce do que outras posicionadas na porção 3’ do mesmo gene. Além disso, houve mais prevalência de aneurismas cerebrais em pacientes com mutações na região 5’ de PKD1.
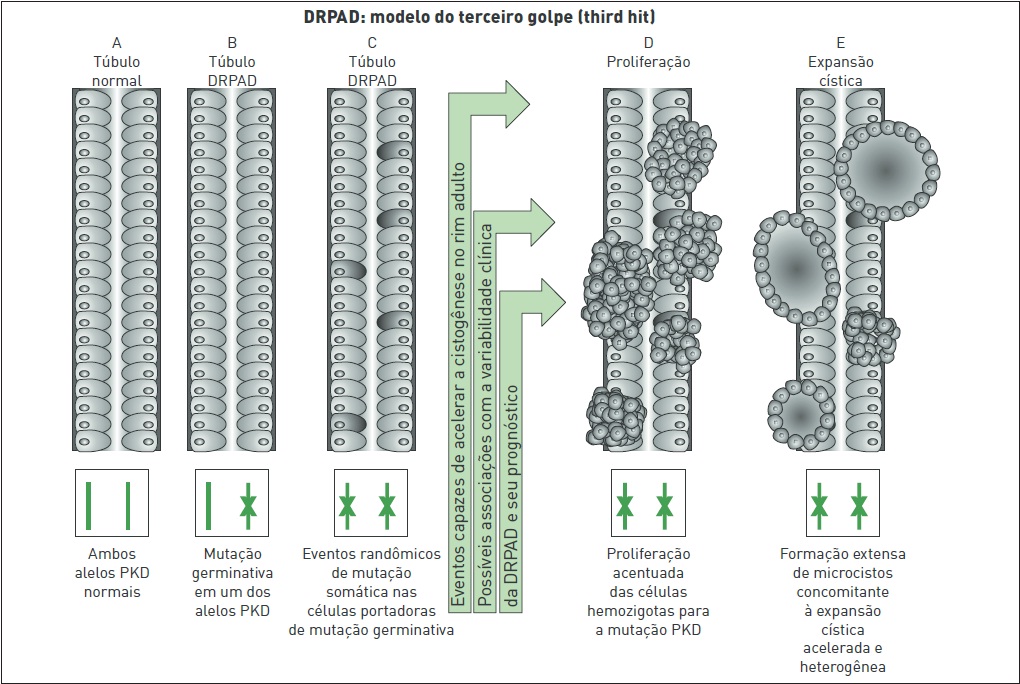
Recentemente, alguns estudos em modelos animais estão fundamentando a hipótese de que haja ainda um terceiro golpe (third hit) envolvido no desenvolvimento da DRPAD (Fig. 88.6).
Figura 88.4
Representação esquemática das policistinas (PC1 e PC2) na membrana celular.
Figura 88.5
Representação esquemática do modelo de dois golpes na formação cística em casos de DRPAD.
De acordo com essa hipótese, a base genética associada a eventos que podem acelerar a cistogênese no rim adulto pode contribuir para a variabilidade clínica da DRPAD e seu prognóstico. Experimentos realizados em modelos animais com isquemia/reperfusão evidenciaram que a lesão isquêmico pode ser considerado como golpe adicional para a formação de cistos renais.
De fato, observações comparativas entre gêmeos univitelinos e irmãos regulares apontam que o desenvolvimento da doença renal é heterogêneo mesmo entre indivíduos com herança gênica semelhante. Outro aspecto importante é o fato de que mecanismos de diferentes naturezas, que podem influenciar a taxa de mutações somáticas sobre células epiteliais tubulares renais, podem potencialmente interferir na gravidade do fenótipo renal, contribuindo também para a variabilidade observada na DRPAD.
Dois aspectos devem ser considerados na formação cística da DRPAD: a proliferação das células tubulares e a secreção de líquido destas. As bases fisiopatológicas para esses eventos dependem, em grande parte, da participação das policistinas 1 e 2 (PC1 e PC2). A interação entre PC1 e PC2 é consistente com a existência de uma via comum que possivelmente é alterada no desenvolvimento da DRPAD.
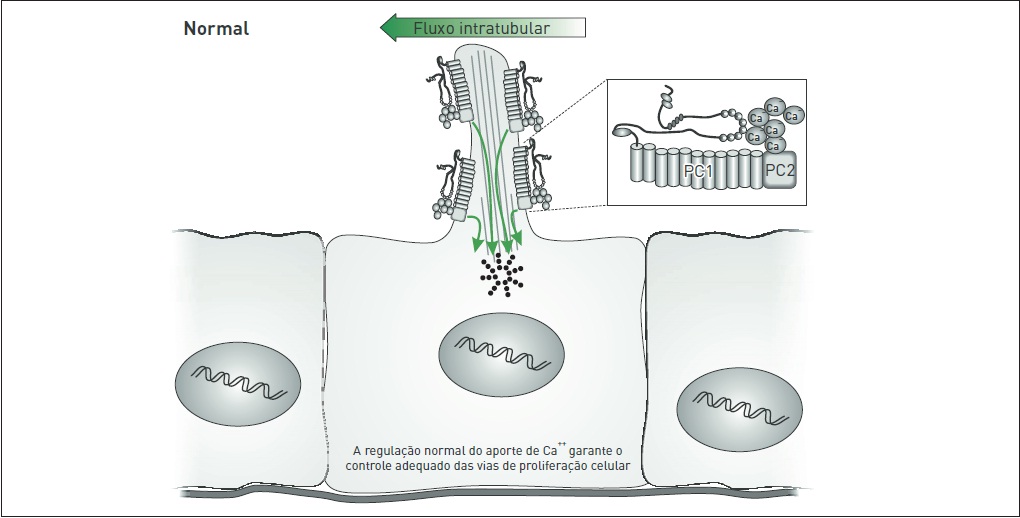
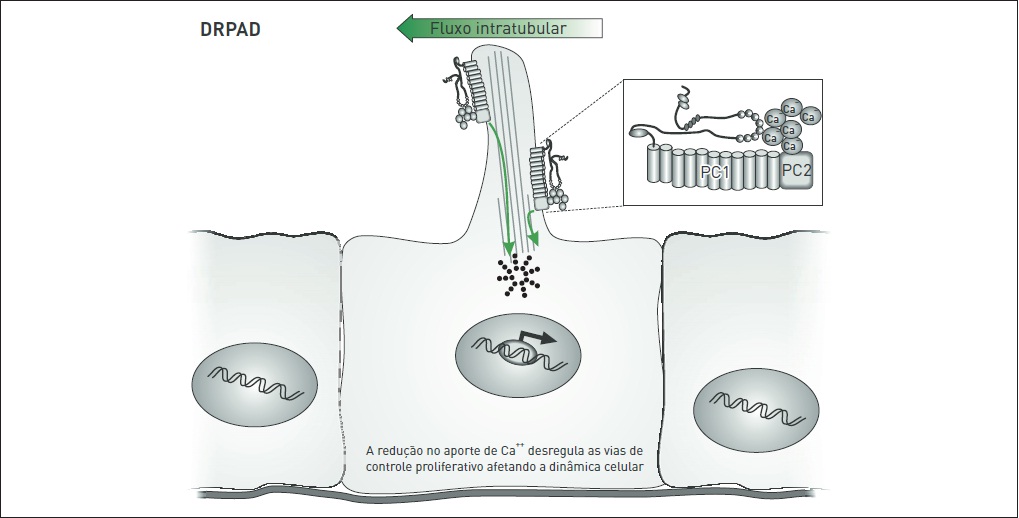
O complexo policistínico (PC1/PC2) localiza-se nos cílios apicais primários de células epiteliais tubulares renais, onde aparentemente age como um mecanossensor que pode identificar estímulos físicos e/ou suas variações, particularmente o fluxo de fluido tubular. Esse processo parece ser mediado pelo influxo de Ca++ por meio de PC2, o que induziria a liberação de Ca++ a partir de estoques intracelulares (Fig. 88.7).
A redução do aporte intracelular de Ca++, observada nas células mutantes para DRPAD, parece determinar o desequilíbrio homeostático desse cátion. Essa alteração, por sua vez, está associada à elevação dos níveis intracelulares de adenosina monofosfato cíclico (AMPc), conforme observado em modelos in vitro. Ao contrário de células renais epiteliais normais, em células DRPAD, o AMPc estimula a via MAPK/ERK (mitogen-ativated protein kinase/extracellular signal-regulated kinase) e induz a proliferação celular (Fig. 88.8). Além disso, observou-se que AMPc pode estimular secreção tran sepitelial de fluido, dirigida por cloreto, em epitélio DRPAD.
Outra relação que parece ser significativa na determinação da gravidade da DRPAD é a proximidade entre os genes PKD1 e TSC2 no braço curto do cromossomo 16. O gene TSC2 é um dos genes mutados em casos de esclerose tuberosa, e o produto de TSC2, a tuberina, parece interagir com a PC1 em um processo que possibilita a ativação da via mamalian target of rapamycin (mTOR) em pacientes com DRPAD.
Figura 88.6
Representação esquemática do modelo de terceiro golpe na formação cística em casos de DRPAD.
Figura 88.7
Infl uxo normal de Ca++ por meio da membrana plasmática, mediado pelo complexo PC1/PC2.
Figura 88.8
Influxo deficiente de Ca++ mediado pelo complexo PC1/PC2 mutado interfere na proliferação celular.
Os sintomas da DRPAD em geral se manifestam em torno de 30 e 40 anos, embora alguns casos apresentem uma expressão clínica significativa já em idade pediátrica (1 a 2%). Diversas manifestações renais e extrarrenais já foram apontadas como complicações importantes.
A diminuição da capacidade de concentração da urina é uma das manifestações mais precoces e pode ser observada em crianças e adolescentes. Nesse contexto, pode ocorrer proteinúria em aproximadamente 20% dos pacientes, mas em geral apresenta nível menor do que 1 g/24 h. A microalbuminúria é mais frequente, ocorrendo em cerca de 35% dos pacientes.
No que se refere à hematúria, observa-se que 30 a 50% dos pacientes, em algum momento durante o desenvolvimento da doença, apresentam perda de sangue na urina, seja ela micro ou macro-hematúria. Outros achados comuns nesses pacientes são sinais e sintomas associados à infecção urinária de repetição. Os cálculos renais também são recorrentes no paciente com DRPAD e podem ser identificados em 20 a 30% dos casos, sendo as composições mais frequentes o oxalato de cálcio e o ácido úrico.
Pode-se identificar nefromegalia ao realizar exame físico, principalmente nos estágios mais avançados da doença.
Nessa situação, os rins podem ser sentidos como massas bilaterais palpáveis e de superfície irregular nos hipocôndrios, geralmente havendo associação com dor em flanco.
Além dos rins, o paciente portador de DRPAD pode apresentar cistos epiteliais no fígado, no pâncreas, no baço, nos pulmões e nos ovários. Esses sinais podem estar associados ou não a prolapso de valva mitral, insuficiência aórtica, divertículos colônicos e aneurismas, sendo esses os principais sinais extrarrenais da DRPAD.
Deve-se ressaltar que outro distúrbio cístico pode ser confundido com a DRPAD em alguns casos. A doença hepática policística autossômica dominante (DHPAD) também é geneticamente heterogênea e apresenta um fenótipo hepático policístico grave, associado ou não à existência simultânea de cistos renais.
A história familiar e os exames de imagem são essenciais para o diagnóstico da DRPAD. Para elaboração da anam nese, deve-se atentar para história familiar de doença cística, com ou sem comprometimento renal. Por apresentar um padrão de herança dominante, espera-se que sejam encontrados membros portadores da DRPAD em todas as gerações. Entretanto, a ocorrência de casos novos deve ser considerada em genealogias em que não seja observado registro prévio de rins policísticos ou doença renal. Não havendo história familiar da doença, o diagnóstico presumível pode ser realizado com evidências de cistos renais bilaterais, de acordo com critérios recentemente unificados por Pei e colaboradores.1 Além disso, a existência de um ou mais dos seguintes critérios também deve ser considerada: aumento bilateral dos rins, cistos hepáticos, pancreáticos ou esplênicos, aneurisma cerebral, cisto solitário aracnoide em glândula pineal e diverticulose.
O exame de imagem apresenta valor determinante para o diagnóstico. Nesse sentido, a ultrassonografia (US) é muito útil no diagnóstico e pode identificar cistos a partir de 1 até 1,5 cm (Fig. 88.1). A existência de cistos hepáticos ou pancreáticos auxilia na confirmação do diagnóstico. Os critérios para diagnóstico ultrassonográfico incluem o número de cistos em cada rim e a idade dos pacientes, conforme a Tabela 88.2. Em termos de sensibilidade, a tomografia computadorizada (TC) é o exame de imagem que pode identificar cistos a partir de 0,5 cm. No entanto, esse exame não é a primeira escolha, seja por utilizar radiação ou por apresentar um custo mais elevado. Por fim, a ressonância nuclear magnética (RM) é considerada um procedimento mais acurado do que a US e deve ser solicitada principalmente nos casos em que a distinção entre carcinoma renal e cistos simples seja necessária. A RM possibilita a verificação de cistos a partir de 3 mm de diâmetro e uma avaliação mais precisa da dimensão dos rins.
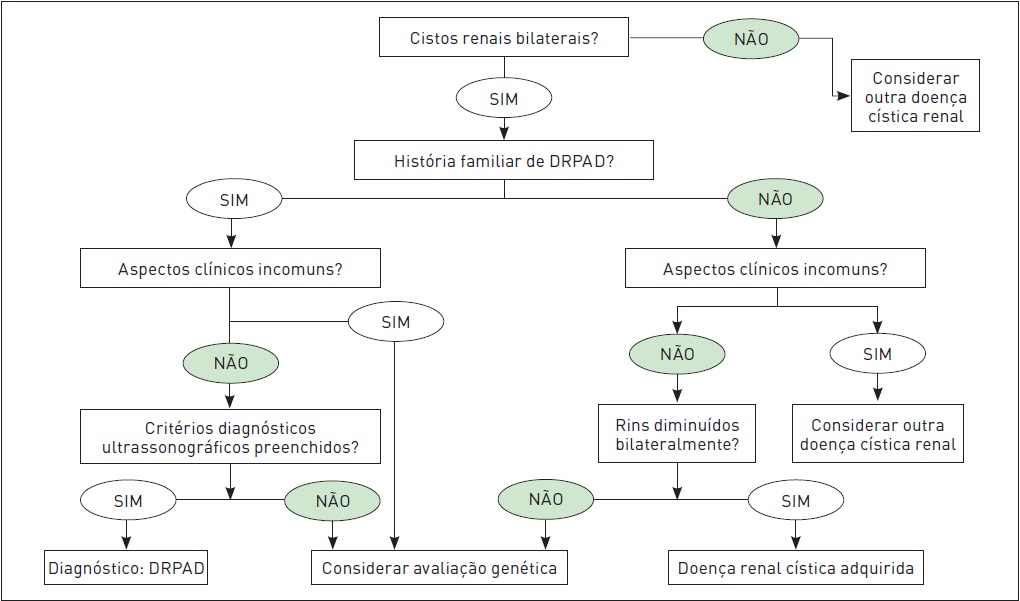
O organograma apresentado na Figura 88.9 ilustra as possíveis etapas do processo diagnóstico da DRPAD.
Figura 88.9
Organograma diagnóstico para DRPAD baseado em informações clínicas e exame de imagem.
O objetivo do tratamento para os pacientes com DRPAD é preservar a função renal e controlar a pressão arterial.
Nesse contexto, deve-se reduzir a progressão para doença renal crônica e monitorar os riscos de ruptura de aneurismas intracerebrais e hemorragia subaracnoide. Outra conduta importante é orientar o paciente a evitar atividades esportivas nas quais haja pos sibilidade de traumas na região lombar ou abdominal, procurando minimizar o risco de ruptura dos cistos.
Em pacientes normotensos e com função renal normal, os exames anuais de função renal e ultrassonografia devem ser periódicos, estabelecendo-se intervalos máximos de 12 meses entre as avaliações. Para o controle da pressão arterial, deve-se utilizar os inibidores da enzima de conversão da angiotensina I (IECAs) ou os antagonistas do receptor da an giotensina II (ARA II), uma vez que o sistema re nina-angiotensina desempenha um papel central na fisiopatologia da hi pertensão nessa situação clínica.
A inibição dos receptores da vasopressina também é um importante desafio como possibilidade terapêutica para a DRPAD, pois o aumento desses receptores pode contribuir diretamente para uma maior concentração de AMPc e interagir com inúmeras outras proteínas associadas à formação cística. Estudos com fármacos específicos para essas condições estão em andamento, e seus resultados poderão auxiliar na conduta clínica em breve.
A dor abdominal é manejada com analgésicos e repouso.
Deve-se evitar o uso de anti-inflamatórios não esteroides devido ao potencial efeito nefrotóxico desses fármacos. Quando os cistos se tornam infectados, o paciente deve ser monitorado e internado. Recomenda-se, nessa situação, administrar antimicrobianos que possam penetrar no cisto, tais como ciprofloxacino, clindamicina, cloranfenicol ou sulfametoxazol-trimetoprima.
A intervenção cirúrgica pode ser necessária nos seguintes casos:
- Dor: a dor aguda pode ser causada por hemorragia intracística ou obstrução renal, seja por coágulo ou litíase. A descompressão do cisto é eficaz no alívio da dor em cerca de 60 a 80% dos casos. Tem-se como opção a drenagem percutânea seguida de instilação de substância esclerosante. Outra possibilidade é a decorticação dos cistos por meio de laparotomia.
- Cistos infectados: esses cistos não são responsivos à antibioticoterapia convencional.
- Nefrectomia: esse procedimento é indicado para pacientes com cistos de grande volume (> 35 cm), infecções recorrentes, hipertensão incontrolável e pos sibilidade de malignidade.
- Doença hepática policística maciça: quando os cistos hepáticos, devido ao grande volume, impossibilitam a realização de uma alimentação adequada ou causam desconforto abdominal grave.
O caso apresentado neste capítulo é um bom exemplo de diagnóstico ocasional de DRPAD, que pode ser precedido de lesão acidental com ruptura cística ou obtido por meio da análise de imagem abdominal de rotina.
Dependendo da disponibilidade de exames de imagem com maior grau de resolução, é possível que cistos em estágios iniciais de desenvolvimento sejam identificados, possibilitando o diagnóstico precoce (Tab. 88.2), o que é fundamental para orientar a conduta clínica. Em casos específicos e havendo disponibilidade, recomenda-se avaliação genética e análise molecular para que sejam identificados outros casos na família que permitam o acompanhamento precoce do paciente.
1. Pei Y, Obaji J, Dupuis A, Paterson AD, Magistroni R, Dicks E, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. 2009;20(1):205-12.
2. Sesso R, Anção MS, Madeira SA. Aspectos epidemiológico do tratamento dialítico na grande São Paulo. Rev Assoc Med Bras. 1994;40(1):10-4.
3. Nunes AC, Milani V, Porsch DB, Rossato LB, Mattos CB, Roisenberg I, et al. Frequency and clinical profile of patients with polycystic kidney disease in southern Brazil. Ren Fail. 2008;30(2):169-73.
4. Iglesias CG, Torres VE, Offord KP, Holley KE, Beard CM, Kurland LT. Epidemiology of adult polycystic kidney disease, Olmsted County, Minnesota: 1935-1980. Am J Kidney Dis. 1983;2(6):630-9.
5. Dalgaard OZ, Soren N. Autosomal dominant polycystic kidney disease in the 1980’s. Clinical Genetics. 1989;36:320-5.
6. Gabow PA. Autosomal dominant polycystic kidney disease. N Engl J Med. 1993;329(5):332-42.
7. 6. Higashihara E, Nutahara K, Kojima M, Tamakoshi A, Yoshiyuki O, Sakai H, et al. Prevalence and renal prognosis of diagnosed autosomal dominant polycystic kidney disease in Japan. Nephron. 1998;80(4):421-7.
8. Glassberg KI. Renal dysplasia and cystic disease of the kidney. In: Walsh PC, Campbell MF, editors. Campbell´s urology. 7th ed. Philadelphia: Elsevier; 1998. p. 1757-813.
9. Hwang YH, Ahn C, Hwang DY, Lee EJ, Eo HS, Chae HJ, et al. Clinical characteristics of end-stage renal disease in autosomal dominant polycystic kidney disease in Koreans. J Am Soc Nephrol. 2000;11:392A.
10. United States Renal Data System [Internet]. Minneapolis: USRDS; c2012 [capturado em 15 set. 2012]. Disponível em: http://www.usrds.org/.
11. Harris PC, Torres VE. Polycystic kidney disease. Annu Rev Med. 2009;60:321-37.
Bastos AP, Piontek K, Silva AM, Martino D, Menezes LF, Fonseca JM, et al. Pkd1 haploinsufficiency increases renal damage and induces microcyst formation following ischemia/reperfusion. J Am Soc Nephrol. 2009;20(11):2389-402.
Braun WE. Autosomal dominant polycystic kidney disease: emerging concepts of pathogenesis and new treatments. Clev Clin J Med. 2009;76(2):97-104.
Bycroft M, Bateman A, Clarke J, Hamill SJ, Sandford R, Thomas RL, et al. The structure of a PKD domain from polycystin-1: implications for polycystic kidney disease. EMBO J. 1999;18(2):297-305.
Cai S, Everitt JI, Kugo H, Cook J, Kleymenova E, Walker CL. Polycystic kidney disease as a result of loss of the tuberous sclerosis 2 tumor suppressor gene during development. Am J Pathol. 2003;162(2):457-68.
Grantham JJ, Torres VE, Chapman AB, Guay-Woodford LM, Bae KT, King BF Jr, et al. Volume progression in polycystic kidney disease. N Engl J Med. 2006;354(20):2122-30.
Pei Y. Pratical genetics for autosomal dominant polycystic kidney disease. Nephron Clin Pract. 2011;118(1):c19-30.
Pei Y, Paterson AD, Wang KR, He N, Hefferton D, Watnick T, et al. Bilineal disease and trans-heterozygotes in autosomal dominant polycystic kidney disease. Am J Hum Genet. 2001;68(2):355-63.
Qian F, Watnick TJ, Onuchic LF, Germino GG. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell. 1996;87(6):979-87.
Rossetti S, Burton S, Strmecki L, Pond GR, San Millán JL, Zerres K, et al. The position of the polycystic kidney disease 1 (PKD1) gene mutation correlates with the severity of renal disease. J Am Soc Nephrol. 2002;13(5):1230-7.
Takakura A, Contrino L, Beck AW, Zhou J. Pkd1 inactivation induced in adulthood produces focal cystic disease. J Am Soc Nephrol. 2008;19(12):2351-63.
Torres V. Vasopressin antagonists in polycystic kidney disease. Semin Nephrol. 2008;28(3):306-17.
Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369(9569):1287-301.
Wilson PD. Polycystic kidney disease. N Engl J Med. 2004;350(2):151-64.
Yamaguchi T, Pelling JC, Ramaswamy NT, Eppler JW, Wallace DP, Nagao S, et al. cAMP stimulates the in vitro proliferation of renal cyst epithelial cells by activating the extracellular signal-regulated kinase pathway. Kidney Int. 2000;57(4):1460-71.
Yu S, Hackmann K, Gao J, He X, Piontek K, Garcia-Gonzalez MA, et al. Essential role of cleavage of Polycystin-1 at G protein-coupled receptor proteolytic site for kidney tubular structure. Proc Natl Acad Sci U S A. 2007;104(47):18688-93.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.