(Carregando Índice)... (Carregando Índice)... |
Autores:
Herlon Saraiva Martins
Médico Assistente do Pronto-Socorro do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (HC-FMUSP) – Disciplina de Emergências Clínicas
Rodrigo Antonio Brandão Neto
Médico Assistente da Disciplina de Emergências Clínicas do Hospital das Clínicas da Faculdade de Medicina da USP
Última revisão: 17/11/2010
Comentários de assinantes: 0
Paciente de 66 anos de idade, sexo masculino, apresenta quadro de tosse seca e dispneia progressiva há 6 meses, não apresenta edema periférico e nega febre e outras manifestações sugestivas de doença sistêmica. Sempre trabalhou como bancário e não apresenta outros antecedentes ocupacionais dignos de nota, também não possui animais em casa.
BEG, pletórico, anictérico, acianótico.
Pressão arterial: 110 x 70 mmHg.
Frequência cardíaca: 100 bpm.
Aparelho respiratório: MV +, com esterotores creptantes teleinspiratórios bibasais em velcro.
Aparelho cardiovascular: 2BRNF, sem sopros.
Abdome: sem alterações dignas de nota.
Membros inferiores: extremidades bem perfundidas, pulsos palpáveis, sem edema periférico.
ECO: FE 60% com pressão de artéria pulmonar de 50mmHg ( normal até 35), sem valvopatias.
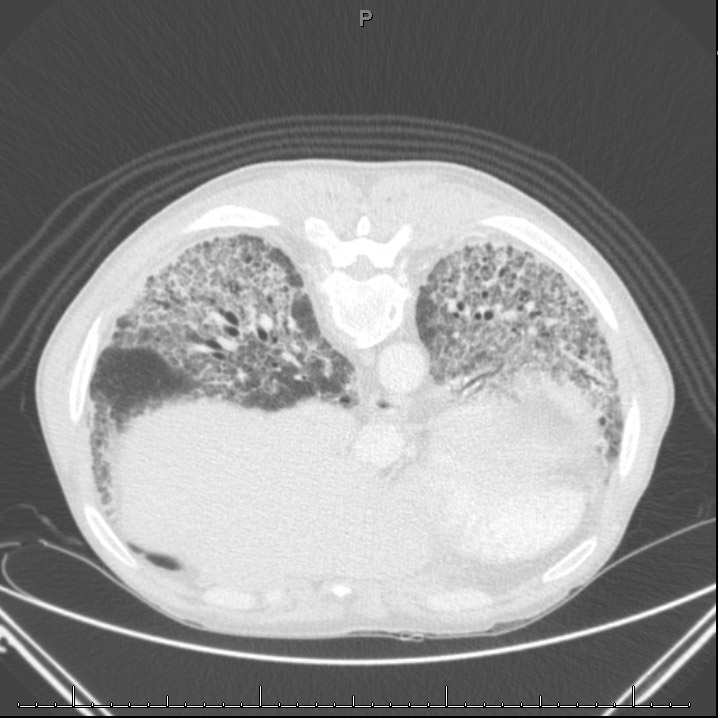
Radiografia de tórax: infiltrado intersticial padrão reticulonodular.
Tomografia de tórax anexa (Figura 1).
Figura 1.

As doenças pulmonares intersticiais (DPI) representam um grande número de condições que envolvem o parênquima pulmonar, ou seja, o alvéolo, a membrana alveolocapilar, o endotélio, as estruturas perivasculares e os linfáticos. Apesar da diversidade de causas, essas doenças são colocadas no mesmo grupo por conta das manifestações fisiopatológicas, clínicas e radiológicas semelhantes. São doenças que acarretam considerável morbimortalidade e existe pouco consenso no manejo ideal delas.
Podem se manifestar apenas com comprometimento pulmonar ou como parte de uma doença sistêmica; podem ter causas conhecidas ou não conhecidas; a doença pode ser aguda, subaguda ou crônica. O processo patológico de base pode ser inflamação com fibrose ou processo granulomatoso. O nosso paciente não tem manifestações sistêmicas, o que sugere um quadro causado por uma doença apenas com comprometimento pulmonar.
Dentre o imenso número de causas, as mais frequentes são:
1. Etiologia não conhecida:
sarcoidose;
pneumonia intersticial fibrosante idiopática (antigamente denominada de fibrose pulmonar idiopática);
doença intersticial associada a doenças do tecido conectivo.
2. Etiologia conhecida:
doenças ocupacionais ou ambientais;
pneumonite de hipersensibilidade;
medicamentosa.
A Tabela 1 cita as principais causas de doenças pulmonares intersticiais não granulomatosas, e a Tabela 2 cita as granulomatosas.
Tabela 1: Doenças pulmonares intersticiais não granulomatosas
|
Causas conhecidas | |
|
Ambientais |
Asbestose, gases tóxicos |
|
Medicamentosas |
Amiodarona, antibióticos, quimioterápicos e sais de ouro |
|
Sequelares |
Pneumonia aspirativa crônica, síndrome do desconforto respiratório do adulto (SDRA) |
|
Causas desconhecidas | |
|
Pneumonias intersticiais idiopáticas |
Pneumonia intersticial fibrosante idiopática (antigamente denominada fibrose pulmonar idiopática); pneumonia intersticial descamativa; pneumonia intersticial associada com bronquiolite; pneumonia intersticial aguda; pneumonia em organização criptogênica (também conhecida como BOOP) |
|
Doenças do tecido conectivo |
Dermatopolimiosite, lúpus eritematoso sistêmico, esclerodermia sistêmica progressiva, artrite reumatoide, espondilite anquilosante, síndrome de Sjögren |
|
Trato gastrintestinal |
Doença inflamatória intestinal, hepatite crônica ativa, cirrose biliar primária |
|
Hereditárias |
Esclerose tuberosa, neurofibromatose, Niemann-pick, Gaucher |
|
Outras |
Amiloidose, proteinose alveolar, pneumonias eosinofílicas, pneumonias intersticiais linfoides etc. |
Tabela 2: Doenças pulmonares intersticiais granulomatosas
|
Causas conhecidas | |
|
Ambientais |
Berílio, sílica e pneumonite de hipersensibilidade |
|
Causas desconhecidas | |
|
Vasculites granulomatosas |
Granulomatose de Wegener e síndrome de Churg-Strauss |
|
Outras granulomatoses |
Sarcoidose, granulomatose das células de Langerhans, granulomatose linfomatoide e granulomatose broncocêntrica |
O mecanismo pelo qual vários agentes diferentes levam à destruição do interstício pulmonar não é completamente conhecido. De maneira geral, após insulto pulmonar (único ou de repetição), a resposta biológica pode ocorrer de duas formas:
1. Processo granulomatoso: ocorre acúmulo de linfócitos T, macrófagos e células epitelioides organizados em estruturas discretas, embora difusamente presentes, denominadas granulomas. As lesões granulomatosas podem evoluir para fibrose pulmonar. A maioria dos doentes permanece pouco sintomática até fases avançadas e costumam melhorar com o tratamento. Além de vasculites sistêmicas, um grande diagnóstico diferencial é feito entre sarcoidose, pneumonite de hipersensibilidade e infecções (tuberculose).
2. Inflamação e fibrose sem granulomas: a injúria é iniciada na superfície epitelial causando inflamação dos espaços aéreos e alvéolos. Uma vez continuado o processo, a inflamação pode estender-se para estruturas adjacentes (interstício, linfáticos e vasculatura), culminando com fibrose intersticial. O desenvolvimento de fibrose avançada é uma condição grave, temida e que pode levar o doente a graves limitações (alterações na troca gasosa e na função ventilatória). Histopatologicamente, os doentes podem ser classificados como:
pneumonia intersticial usual (pneumonite fibrosante ou fibrose idiopática);
pneumonia intersticial inespecífica;
predomínio de bronquiolite respiratória;
pneumonia em organização criptogênica (também conhecida como BOOP – pneumonia com bronquiolite obliterante em organização);
pneumonia intersticial descamativa;
infiltração linfocítica.
Os doentes podem procurar assistência médica em virtude de sua própria doença de base (p.ex., lúpus, polimiosite, doença inflamatória intestinal etc.) ou pelo surgimento de sintomas respiratórios (dispneia, tosse, sibilância, hemoptise, dor torácica etc.). Alguns dados importantes são descritos a seguir.
Dispneia costuma ser a principal queixa referida pelo doente. De início, ocorre aos esforços, em razão da dificuldade de troca de oxigênio pela barreira à difusão imposta pela doença intersticial, entretanto, pode evoluir com dispneia em repouso, ortopneia e mesmo dispneia paroxística noturna (DPN). Dor torácica não é comum; o súbito surgimento de dor tipo pleurítica deve sugerir complicação com pneumotórax. Escarro com laivos hemoptoicos e hemoptise podem ocorrer.
Fadiga, perda de peso e anorexia são comuns.
As manifestações clínicas podem surgir de qualquer forma, em uma escala temporal, citada na Tabela 3. Entretanto, a forma mais frequente é a apresentação insidiosa e crônica.
Tabela 3: Padrão temporal e causas mais frequentes
|
Início dos sintomas |
Comentários |
|
Agudo (dias a semanas) |
Pneumonite de hipersensibilidade, pneumonia eosinofílica, pneumonia intersticial idiopática aguda e reações imunológicas (medicamentos, helmintos, fungos). Um grande diagnóstico diferencial é com pneumonia bacteriana de apresentação intersticial (Mycoplasma, Legionella, Chlamydia etc.) |
|
Subagudo (semanas a meses) |
Pode ocorrer com todas as DPI. Mais frequente com a sarcoidose, pneumonia intersticial induzida por medicamentos, pneumonia em organização criptogênica (também conhecida como BOOP) e formas intersticiais associadas a doenças do tecido conectivo (especialmente lúpus e polimiosite). |
|
Crônico (vários meses) |
Globalmente, é forma mais comum de apresentação. O protótipo é a pneumonia intersticial fibrosante idiopática. |
Em geral, há duas grandes faixas etárias: após 50 anos de idade (pneumonia intersticial fibrosante idiopática) e entre os 20 e 40 anos (histiocitose, sarcoidose, formas familiares ou doenças genéticas). Doenças do tecido conectivo também são mais comuns em mulheres, exceto vasculite reumatoide (mais comum em homens). A maioria das outras doenças, inclusive as ocupacionais, é mais comum em homens.
Essa pesquisa deve ser exaustiva e detalhada, inclusive com amigos e familiares. Várias formas de exposição crônica podem levar à DPI (asbesto, sílica, gases, berílio etc.). Outras vezes, a exposição a determinado agente é seguida de um quadro clínico de febre, calafrios, tosse, sibilos e dispneia. Novas exposições podem ocasionar quadros semelhantes (p.ex., agricultor, fazendeiro, criador de pombos etc.).
Quase 75% dos doentes com pneumonia intersticial fibrosante idiopática (PIFI) têm história de tabagismo. Quase todos os doentes com histiocitose, pneumonia intersticial descamativa, bronquiolite respiratória, síndrome de Goodpasture e proteinose alveolar têm história de tabagismo.
Taquipneia é comum e os doentes podem ter respirações rápidas e superficiais. Crepitações em ambas as bases, eventualmente difusas (“crepitações secas”) são comuns e podem ser audíveis, assim como roncos difusos. Sibilos são raros, mas podem ocorrer na síndrome de Churg-Strauss, pneumonite de hipersensibilidade, pneumonias eosinofílicas, bronquiolite respiratória e sarcoidose. Sinais de hipertensão pulmonar podem surgir em fases mais avançadas. Baqueteamento digital e cianose podem surgir. Dados da doença de base também podem predominar (p.ex., artrite, rash malar, vasculite de pele, linfonodomegalias, eritema nodoso etc.).
Podem ser solicitados de acordo com a hipótese clínica. Em geral, pelo menos hemograma, eletrólitos, função renal, exames de coagulação, urina tipo 1, eletrocardiograma (ECG) e radiografia de tórax são solicitados. Uma medida da oximetria de pulso deve ser feita e uma gasometria arterial deve ser colhida. A gasometria pode ser normal em repouso, mas mostrar hipoxemia após exercício; ela pode mostrar hipoxemia em repouso e, na sequência, hipoxemia com hipocapnia, hipoxemia grave com hipercapnia (indicando estádio avançado da doença).
Vários cenários clínicos podem alterar os exames laboratoriais, como lúpus (anemia, leucopenia, plaquetopenia, lesão renal etc.), vasculites sistêmicas (função renal alterada, hipercalemia etc.). Outros exames podem ser solicitados, como:
fator antinúcleo e outros autoanticorpos: podem indicar lúpus (FAN positivo, anti-Sm, anti-DNA de dupla fita), esclerodermia (anti-Scl 70 ou anti-topoisomerase III), polimiosite (anti-Jo 1 ou anti histidil t-RNA sintetase) etc.;
fator reumatoide: positivo em várias doenças. Altos títulos são comuns na vasculite reumatoide;
anticorpo antiproteinase 3 (cANCA): também denominado anticorpo anticitoplasma de neutrófico padrão citoplasmático. Indica granulomatose de Wegener;
anticorpo antimieloperoxidase (pANCA): também denominado anticorpo anticitoplasma de neutrófilo padrão perinuclear. Pode ser positivo em várias doenças (hepatite crônica ativa, poliangeíte microscópica), inclusive, na síndrome de Churg-Strauss e na granulomatose de Wegener;
anticorpo antimembrana basal glomerular (domínio não colagenoso do colágeno tipo IV): indica Goodpasture;
enzimas musculares: aumentadas significativamente, podem sugerir polimiosite;
desidrogenase lática: inespecífica; costuma aumentar na maioria das DPI;
dosagem da enzima conversora de angiotensina: pode ocorrer na sarcoidose;
cálcio sérico: pode ocorrer na sarcoidose.
Costumam indicar um distúrbio restritivo: capacidade vital forçada (CVF) diminuída, volume expiratório forçado de 1º segundo (VEF1) e relação VEF1/CVF normais ou aumentados.
Medidas diretas de volumes pulmonares confirmam uma doença restritiva, mostrando uma redução importante da capacidade pulmonar total (CPT). Muito raramente, algumas doenças podem cursar com distúrbio combinado ou misto (restritivo e obstrutivo), tais como a linfangioleiomiomatose e a esclerose tuberosa (mais raro: sarcoidose e pneumonite de hipersensibilidade).
A redução na capacidade de difusão do monóxido de carbono (DLCO) é muito comum, embora não específico de DPI. Embora pareça mais lógico pensar que a redução da difusão se deva a “espessamento” do espaço entre o alvéolo e o capilar, o mais importante determinante da redução do DLCO é o desequilíbrio ventilação-perfusão (distúrbio V/Q). Isso ocorre porque as regiões infiltradas e fibróticas apresentam redução da ventilação.
É importante notar que a gravidade da redução da DLCO não se correlaciona tão bem quanto a gravidade da doença.
Um grande aumento da DLCO pode significar sangue no interstício pulmonar e ocorre em síndromes de hemorragia alveolar.
Mesmo com grande acometimento pulmonar, doentes com DPI podem apresentar gasometria de repouso normal. Entretanto, quando o doente é induzido a exercitar-se, ele deve recrutar uma parte do espaço morto, com intuito de aumentar a troca gasosa (a fração de espaço morto normal é menor que 0,4).
Normalmente, o espaço morto consiste de duas formas: espaço morto anatômico e espaço morto fisiológico ou funcional. O espaço morto anatômico é aquela porção do volume corrente que não entrou em contato com alvéolos perfundidos e, por isso, não participa da troca gasosa. Já o espaço morto funcional ou fisiológico é a soma do excesso de ventilação em relação à perfusão (ou seja, locais com muito ar e pouco sangue), que são áreas onde o V/Q é maior que 1.
Um volume fixo de gases é requerido, em cada respiração, para preencher o espaço morto anatômico (gases que estão em vias aéreas proximais até bronquíolos). Por isso, em geral, durante testes com esforço físico, doentes com DPI podem não conseguir recrutar esse espaço morto funcional, compensando por meio do aumento da frequência respiratória, na prática, reduzindo o volume corrente (VT). O que acontece é o desenvolvimento de hipoxemia com hipocapnia ao esforço.
Medidas seriadas de testes ergoespirométricos podem ser úteis no seguimento de doentes com DPI.
A radiografia de tórax é o exame que faz a suspeita inicial de DPI. Muitas vezes, os achados radiológicos são inespecíficos e duvidosos. Doentes com DPI podem ter radiografia de tórax normal. Entretanto, quando alterada, o acometimento pulmonar pode ocorrer com as seguintes formas:
infiltrado reticular bibasal ou infiltrado reticular e nodular (mais frequentes);
opacidades nodulares predominantemente em campos pulmonares superiores: sarcoidose, histiocitose, pneumonite de hipersensibilidade, silicose, beriliose, vasculite reumatoide nodular;
padrão em favo de mel (honeycomb) indica múltiplos pequenos espaços císticos com fibrose e se correlaciona com doença avançada.
Além de definir a extensão da doença, ela pode mostrar alterações torácicas adicionais (p.ex., gânglios hilares, mediastinais, câncer, linfangite carcinomatosa, enfisema etc.). Muitas vezes, o achado da TCAR é suficiente para diagnosticar pneumonia intersticial fibrosante idiopática, sarcoidose, asbestose, pneumonite de hipersensibilidade e histiocitose das células de Langerhans (Tabela 4). Pode também ser útil no seguimento dos doentes (resposta à terapia) e delinear o melhor local para biópsia pulmonar.
Tabela 4: Métodos de imagem e diagnóstico específico
|
Achados |
Considerações diagnósticas |
|
Adenopatia hilar* |
Sarcoidose, infecções granulomatosas, linfoma, linfangite carcinomatosa |
|
Derrame pleural |
Doenças do tecido conectivo, asbestose, infecções |
|
Pneumotórax |
Histiocitose, linfangioleiomiomatose, infecções |
|
Predomínio de campos superiores |
Silicose, sarcoidose, histiocitose. |
|
Predomínio de zonas periféricas |
Medicamentosa, pneumonia eosinofílica, pneumonia em organização criptogênica (BOOP). |
* Também pode sugerir diagnósticos diferenciais: linfomas, carcinomas.
Um diagnóstico de certeza propicia algumas vantagens:
permite diagnosticar doenças “tratáveis”: sarcoidose, pneumonite de hipersensibilidade crônica, pneumonia em organização criptogênica (também conhecida como BOOP) etc.;
evita confusão e ansiedade, especialmente quando não há resposta terapêutica;
evita terapêuticas potencialmente tóxicas em situações em que ela não é indicada.
Há duas maneiras de se obter tecido pulmonar para estudo anatomopatológico; ambas são descritas a seguir.
É a mais indicada inicialmente. A maior vantagem da biópsia transbrônquica é o menor risco para o doente. A maior desvantagem é a incapacidade de fornecer um diagnóstico definitivo na maioria das vezes. As situações onde ela pode ser mais útil são: sarcoidose, pneumonite de hipersensibilidade, pneumonia eosinofílica, proteinose alveolar, infecções.
Pode ser feita por videotoracoscopia (mais indicada) ou por toracotomia. Em geral, é feita após uma transbrônquica ser inespecífica. Obviamente, acarreta risco ao doente (inerente a anestesia, fístula pleural, infecção, sangramento, óbito).
O curso clínico e o tratamento das DPI é altamente variável e, em parte, depende da causa de base. Todas as causas potencialmente tratáveis devem ser buscadas antes que fibrose sequelar ocorra. Por isso, é importante identificar doenças que possam ter alguma resposta benéfica com corticoides, descritas na Tabela 5. Os achados clínicos das principais doenças intersticiais estão descritos nas Tabelas 6 e 7.
Tabela 5: Doenças que podem responder bem a corticoides
|
Pneumonias eosinofílicas |
Pneumonia em organização criptogênica (bronquiolite obliterante com pneumonia em organização) |
|
Sarcoidose |
Doenças do tecido conectivo |
|
Pneumonites de hipersensibilidade |
Pneumonite aguda associada à radiação |
|
Síndromes de hemorragia alveolar |
Pneumonites associadas medicamentos |
Tabela 6: Características das doenças intersticiais
|
|
Sarcoidose |
Pneumonia intersticial descamativa |
Pneumonia intersticial linfocítica |
Doenças do tecido conectivo |
|
Achados clínicos |
Mulheres negras; sintomas mínimos; lesões cutâneas |
Homens de idade média, tabagistas; dispneia progressiva |
Mulheres idosas com doenças associadas |
Assintomáticos; pode evoluir com dispneia progressiva, tosse, dor torácica, hemoptise |
|
Radiografia de tórax |
Adenopatia hilar bilateral; predomínio de campos superiores |
Predomínio bibasal; pode ser normal |
Reticulonodular |
Infiltrados bilaterais, difusos ou focais |
|
Tomografia computadorizada de cortes finos |
Adenopatia hilar e mediastinal. Nódulos, distribuição perilinfática e broncovascular |
Fibrose com distorção alveolar. Padrão vidro despolido |
Padrão vidro despolido, nódulos centrilobulares, espessamento septal e broncovascular |
Fibrose com ou sem vidro despolido |
|
Achados extratorácicos |
Exuberante |
Nenhum |
Depende da doença associada (HIV, transplante, autoimune, drogas etc.) |
Exuberante e depende da doença autoimune de base |
|
Patologia |
Granulomas não caseosos |
Preenchimento homogêneo peribronquiolar |
Infiltrado das paredes alveolares com linfócitos |
Variável: inclui destruição alveolar difusa, bronquiolite etc. |
|
Terapia |
Maioria não necessita de tratamento. Boa resposta a corticoide |
Parar de fumar e corticoide |
Tratar a doença de base; boa resposta a corticoides |
Tratar a doença de base; em geral, necessita de corticoide e imunossupressor |
|
Prognóstico |
Excelente |
Sobrevida de 12 anos; mortalidade de 20 a 30% |
Depende da doença de base |
Depende da doença de base |
Tabela 7: Características das doenças intersticiais
|
|
Granuloma eosinófilo |
Proteinose alveolar |
Pneumonia eosinofílica |
Linfangioleiomiomatose |
|
Achados clínicos |
Idade de 20 a 50 anos; insidioso com tosse e dispneia; tabagistas |
Tosse e dispneia progressivos |
Mulher jovem; tosse, febre, dispneia, sibilos |
Mulher em idade de procriação; dispneia, hemoptise, pneumotórax, quilotórax |
|
Radiografia de tórax |
Nódulos, cistos, pneumotórax, derrame pleural |
Preenchimento alveolar bibasal (semelhante edema pulmonar, mas sem congestão) |
Infiltrado bilateral |
- |
|
Tomografia computadorizada de cortes finos |
Cistos em formas bizarras, com nódulos disseminados |
Opacidades em vidro despolido, espessamento dos septos interlobulares |
Padrão vidro despolido bilateral |
Cistos de paredes finas, tamanhos variados, dilatação do ducto torácico, derrame pericárdico e pleural, pneumotórax |
|
Achados extratorácicos |
Lesões ósseas e diabetes insípido |
Não |
Eosinofilia |
Angiomiolipomas renais |
|
Patologia |
Lesões nodulares contendo células de Langerhans com grânulos de Birbeck |
Material alveolar, eosinofílico com coloração positiva para o PAS |
Intenso infiltrado misto, rico em eosinófilos |
Proliferação difusa de células musculares lisas com formação de cistos |
|
Terapia |
Parar de fumar |
Lavagem pulmonar total |
Boa resposta a corticoides |
Progesterona e/ou ooforectomia |
|
Prognóstico |
Péssimo; sobrevida de 6 anos |
Bom prognóstico |
Bom prognóstico |
Mortalidade de 25% em 8 anos |
O primeiro passo é tentar afastar doenças ou condições que possam simular uma doença pulmonar intersticial (DPI), tais como:
1. Obesidade: pode cursar com dispneia, espirometria com padrão restritivo e a radiografia de tórax pode falsamente sugerir infiltrado intersticial (aumento de partes moles).
2. Linfangite carcinomatosa: quadro clínico subagudo (semanas a poucos meses) com tosse, dispneia, taquipneia, espirometria com padrão restritivo, radiografia com achados variados (pode cursar com infiltrado intersticial bilateral, simétrico ou não, nodulações etc.). A tomografia de tórax costuma sugerir o diagnóstico, mostrando um envolvimento pulmonar linfático. Em geral, cursa com um péssimo prognóstico, podendo ser causada por câncer primário de pulmão ou câncer metastático.
3. Edema pulmonar cardiogênico: sinais e sintomas de ICC, derrame pleural bilateral, área cardíaca aumentada etc.
4. Medicações: vários medicamentos podem causar infiltrado intersticial. Se houver suspeita, deve-se suspendê-lo imediatamente. A Tabela 8 cita as principais medicações associadas a presença de infiltrado intersticial.
Tabela 8: Medicações associadas a infiltrado intersticial
|
Quimioterápicos |
Bleomicina |
Dose-relacionada |
|
Bussulfam |
Fibrose pulmonar; pode cursar com proteinose alveolar | |
|
Ciclofosfamida |
Curso muito variável | |
|
Vimblastina |
Sinérgico com mitomicina-C | |
|
Nitrosureias |
Início retardado e dose-relacionado | |
|
Cardiovasculares |
Amiodarona |
Longa meia-vida; pode causar SARA e pneumonite de hipersensibilidade |
|
Hidralazina |
Síndrome lúpus-símile | |
|
Anti-inflamatórios |
Metotrexato |
Pode produzir granulomas |
|
Penicilamina |
Síndrome lúpus-símile, pneumonia em organização criptogênica (também conhecida como BOOP) | |
|
Antibióticos |
Nitrofurantoína |
DPI aguda ou crônica; variável curso clínico |
|
Sulfassalazina |
Fibrose, pneumonia em organização criptogênica (também conhecida como BOOP), pneumonite intersticial |
O nosso paciente apresenta dados epidemiológicos como idade e achados clínicos que sugerem o diagnóstico de fibrose pulmonar idiopática ou de pneumonia intersticial fibrosante idiopática. Doentes com ausência de exposição ambiental, ocupacional, uso de medicamentos, doenças autoimunes, em geral, apresentam uma pneumonia intersticial fibrosante idiopática (PIFI), anteriormente denominada fibrose pulmonar idiopática.
Um passo importante no diagnóstico de PIFI é excluir as causas conhecidas. Em geral, a doença é confirmada pelo anatomopatológico. As principais formas da PIFI, bem como os achados clínicos, anatomopatológico, radiológico, tratamento e prognóstico estão descritos na Tabela 9.
A fibrose pulmonar idiopática (FPI) é uma doença de caráter progressivo e insidioso com sobrevida média, a partir do momento do diagnóstico, de 4 a 6 anos. Uma das suas principais características é o desenvolvimento de infiltrados intersticiais com predomínio em bases pulmonares com dispneia progressiva e piora da função pulmonar.
Patologicamente, caracteriza-se por zonas fibróticas como favos de mel associadas a áreas de tecido relativamente não afetadas. Em geral, o processo inflamatório é relativamente leve, considerando a extensão da doença; já a doença intersticial pulmonar com bronquiolite usualmente associa-se com espessamento relativamente uniforme dos bronquíolos do septo alveolar e acúmulo de macrófagos intra-alveolares.
O insulto que leva à doença pulmonar não é identificado, mas inicia um processo de inflamação crônica do tipo Th2. Cerca de 75% dos pacientes com doença tem antecedente de tabagismo, e pode ainda haver associação com infecções por alguns herpes-vírus.
A apresentação típica é de dispneia aos esforços com tosse seca, que aparece usualmente na 5ª a 6ª década de vida e é mais frequente em homens que em mulheres. Sintomas associados incluem febre baixa e mialgias, que são incomuns e, em raros casos, podem preceder o aparecimento de dispneia. Os pacientes apresentam estertores bibasilares teleinspiratórios em velcro. O baqueteamento digital é descrito em cerca de 50% dos pacientes.
Anormalidades laboratoriais nestes pacientes são inespecíficas; alguns pacientes podem apresentar leve anemia, enquanto outros podem ser pletóricos em razão da hipoxemia crônica; marcadores inflamatórios podem estar aumentados. Fator reumatoide e FAN são descritos positivos em até 30% dos pacientes que não apresentam LES e artrite reumatoide associadas.
A prova de função pulmonar revela, nestes pacientes, distúrbio restritivo com diminuição de capacidade pulmonar total, capacidade funcional residual e volume residual decorrente da diminuição da complacência pulmonar.
Radiologicamente, os pacientes apresentam opacidades bilaterais reticulares, sobretudo em periferia e bases pulmonares, sendo que virtualmente todos os pacientes apresentam radiografia anormal. Em TC, pode-se observar fibrose progressiva com dilatação cística dos espaços aéreos vistos como “favos de mel”, principalmente subpleurais. As anormalidades reticulares são salteadas com espessamento septal, e bronquiectasias de tração são comuns também. Achados como derrame pleural, broncograma aéreo, sombras confluentes ou adenopatia hilar sugerem fortemente outros diagnósticos.
Para pneumologistas experientes, alguns casos podem ter seu diagnóstico baseado apenas na história e no achado tomográfico, porém, em mais de 50% dos casos, é necessário realizar biópsia para diagnóstico definitivo.
A história natural é de uma doença progressiva, com piora da fibrose e diminuição de complacência pulmonar com aumento progressivo do esforço para respirar. Infecções respiratórias baixas são comuns, pois os pacientes apresentam bronquiectasias de tração, dificuldade de clearance de muco, também incidência maior de refluxo gastroesofágico pode ocorrer nestes pacientes.
Disfunção ventricular esquerda ocorre em menos de 10% dos pacientes. Quando presente, está, na maioria das vezes, associada a insuficiência cardíaca direita. Também ocorre uma incidência maior de carcinoma broncogênico. A sobrevida média é de 3 anos, mesmo na ausência de outras doenças complicadoras.
Educação e tratamento de suporte são fatores importantes no tratamento destes pacientes, com oxigenoterapia suplementar e reabilitação pulmonar; também deve-se sugerir a realização de vacinação para pneumococo e influenza.
Nenhum tratamento farmacológico até hoje mostrou benefício claro em alterar o curso da doença. Entretanto, dado o mau prognóstico da doença, recomenda-se o uso de corticoide e/ou drogas imunossupressoras sempre que não houver contraindicações. O tratamento da PIFI baseia-se na hipótese inflamatória da doença, com expectativa de que a resolução do componente inflamatório possa prevenir a subsequente formação/progressão de fibrose.
Glicocorticoides podem apresentar resposta em 10 a 40% dos pacientes. Resposta radiográfica com diminuição da opacidade em vidro despolido pode ocorrer, mas o efeito na evolução da doença é relativamente pequeno. Parece razoável realizar um trial por 3 a 6 meses com glicocorticoide e observar se ocorre melhora em parâmetros fisiológicos; caso negativo, deve-se considerar a retirada da medicação. Terapia combinada com glicorticoide, azatioprina e n-actilcisteína é sugerida pela literatura, em um estudo em que a n-acetilcisteína 600 mg por 3 vezes/dia, combinado a prednisona e azatioprina, foi superior ao uso dos imunossupressores isoladamente, com diminuição da velocidade da queda da capacidade vital e da DLCO. A dose típica do corticoide é prednisona 0,5 mg/kg de peso e a azatioprina em geral dose de 2 mg/kg de peso. Em geral, a terapia combinada é mantida por 8 semanas e, depois, o paciente é reavaliado.
A colchicina era utilizada por seu efeito antfibrótico, por diminuir o processamento intracelular de colágeno, aumentar a expressão das enzimas degradadoras de colágeno e suprimir a liberação pelos macrófagos por fatores e crescimento de fibroblasto. Não existem grandes evidências de seu benefício e, portanto, não recomendamos seu uso.
Outros agentes fibróticos têm sido sugeridos, como a pirferidona, que inibe a produção de síntese de colágeno in vitro. Os estudos com a medicação só mostraram benefício em desfechos secundários, embora pareça funcionar para fibrose associada à síndrome de Hermansky-Pudlak que cursa com albinismo oculocutâneo e alterações plaquetárias. O uso de antagonistas de receptores da endotelina, como o bosentam, também está sendo estudado recentemente e um estudo recente demonstrou sucesso em desfechos secundários, com o uso do sildenafil; também as estatinas estão sendo avaliadas nestes pacientes. A associação da doença com tromboembolismo venoso foi avaliada e alguns autores sugerem que anticoagulação pode ser benéfica com diminuição de mortalidade em um pequeno estudo.
Em algumas séries, pode ocorrer refluxo gastroesofágico em até 90% dos pacientes com FIP; o tratamento adequado pode auxiliar o controle da doença.
Outras medicações avaliadas incluem interferon, ciclofosfamida, imatinibe, ciclosporina, etanercept, penicilamina e ciclosporina, mas ainda não existem evidências para seu uso.
Exacerbações agudas podem ocorrer por infecções, tromboembolismo, pneumotórax ou disfunção cardíaca e, por vezes, idiopática; são associadas com piora importante de prognóstico e os critérios para sua definição incluem:
diagnóstico de fibrose pulmonar idiopática;
aparecimento ou piora da dispneia nos últimos 30 dias;
TC de alta resolução com anormalidades novas com infiltrado em vidro opaco ou consolidações ou padrão em “favo de mel”;
sem evidência de infecção respiratória em aspirado traqueal ou lavado broncoalveolar;
exclusão de diagnósticos alternativos.
Na presença destes cinco critérios, temos uma exacerbação suspeita, e pacientes com novas alterações tomográficas de padrão periférico apresentam melhor prognóstico. O tratamento inclui antibióticos de amplo espectro e corticoterapia com prednisona 1 mg/kg/dia por via oral ou metilprednisolona 1 a 2 mg/kg EV. Pode ser necessária ventilação invasiva, mas a taxa de insucesso é alta, chegando a 78%; recorrência é comum.
Tabela 9: Características da pneumonia intersticial fibrosante idiopática
|
Nome |
Apresentação clínica |
Patologia e radiografia |
Terapia e prognóstico |
|
Pneumonia intersticial usual (piu) |
Idade de 55 a 60 anos; predomínio discreto em homens; início insidioso de tosse e dispneia durante anos; baqueteamento digital em até 50% dos casos; Fan e FR positivos em até 25% dos casos (doença autoimune ausente) |
Distribuição não uniforme de fibrose focal, padrão em “favo de mel” e pulmão normal; tomografia computadorizada de cortes finos mostra doença bilateral, variáveis graus de vidro despolido e “favo de mel” |
Estudos não mostram melhora com tratamento; doença progressiva; cerca de 15% de resposta a citotóxicos e corticoides; média de sobrevida de 3 anos; estudos com antifibrosantes em andamento |
|
Doença intersticial associada com bronquiolite |
Início entre 40 a 45 anos de idade, semelhante à PIU. Todos são tabagistas. |
Arquitetura alveolar preservada; infiltrado abundante de macrófagos; peribronquilar. É raro encontrar fibrose ou padrão “favo de mel”. Tomografia comutadorizada de cortes finos pode mostrar predomínio de vidro despolido. |
Parar de fumar é essencial. Cerca de ¼ dos doentes têm remissão espontânea. Sobrevida maior que 10 anos. Indica-se corticoide, apesar da ausência de estudos. |
|
Pneumonite intersticial aguda |
Início em qualquer idade (jovens a idosos). Curso clínico indistinguível da SARA. Quadro agudo de dispneia e insuficiência respiratória. |
Predomínio de alveolite difusa. Mínima fibrose, deposição de colágeno. Tomografia computadorizada de cortes finos com preenchimento alveolar difuso com vidro despolido. |
Suporte clínico. Alta mortalidade (50 a 90% em 2 meses). Não é progressiva, caso o doente não morra. |
|
Pneumonite intersticial inespecífica |
Início entre 45 a 55 anos de idade. Predomínio discreto em mulheres. Quadro clínico com evolução em meses. |
Achados patológicos variáveis (não se enquadra nas outras formas). Pode ser indistinguível da PIU. Tomografia computadorizada de cortes finos com áreas bilaterais de vidro despolido. Raro encontrar, ao diagnóstico, padrão em “favo de mel” |
Boa resposta ao tratamento, embora, não existam estudos adequados. Sobrevida maior que 10 anos. Corticoides e/ou citotóxicos indicados. |
|
Pneumonia em organização criptogênica (BOOP) |
Início entre 50 e 60 anos de idade (mas pode variar). Início agudo a subagudo (semanas a poucos meses) com dispneia, tosse seca. Sintomas sistêmicos são comuns (fadiga, febre, perda de peso). Espirometria com restrição; obstrução em ¼ dos casos. |
Infiltrado inflamatório com preenchimento de alvéolos e bronquíolos distais. Tomografia computadorizada de cortes finos mostra consolidações periféricas (subpleurais), vidro despolido, espessamento e dilatação da parede bronquiolar. |
Bom prognóstico com tratamento. Recidivas são comuns. Rápida resposta aos oicorticoides (mais de 2/3 dos doentes). |
Os corticoides já foram bastante detalhados em outras discussões de prescrição clínica. Nas doenças intersticiais, entretanto, as doses e a duração do tratamento podem ser bem maiores, acarretando muito mais efeitos colaterais. Outra diferença é que praticamente não há estudos randomizados de qualidade mostrando inequivocamente benefício.
Pneumonia intersticial fibrosante. Apesar do seu uso disseminado, não há ensaios clínicos prospectivos, randomizados, duplo-cegos, placebo-controlados que tenham avaliado o papel dos esteroides na FIP. Em três estudos publicados que compararam o uso de corticoides com placebo, nenhum dos pacientes sem tratamento apresentou melhora. Em uma revisão recente da Cochrane, foram selecionados cerca de 15 estudos que incluíram pacientes com fibrose pulmonar e foram tratados com corticoides. A conclusão é que não há dados a favor ou contra o uso de corticoides, haja vista que todos os estudos tinham graves erros metodológicos.
Mesmo consideradas as limitações dos estudos existentes, a potencial melhora com uso de corticoide ocorre somente em 10 a 30% dos pacientes e tende a ser transitória. Habitualmente, a dose utilizada é de 40 a 100 mg de prednisona (ou equivalente) por via oral por 2 a 4 meses com desmame progressivo. A melhora que ocorre com o tratamento geralmente se manifesta nos primeiros 3 meses. Portanto, a prática clínica corrente é obter parâmetros clínicos objetivos ao final de 3 meses (prova de função pulmonar, tomografia de cortes finos e escalas de dispneia) para decidir sobre manutenção ou suspensão do tratamento. Deve-se manter o tratamento somente se o paciente apresentar melhora ou estabilização do quadro. Naqueles pacientes respondedores, deve-se manter o tratamento com esteroides cronicamente em doses progressivamente menores. Qualquer sinal de piora clínica com o desmame do corticoide sugere necessidade de aumento da dose ou de associação com imunossupressores.
A azatioprina é convertida à mercaptopurina, um análogo purínico que afeta a síntese de DNA e RNA. A imunidade celular é afetada de forma mais importante que a humoral. O mecanismo exato pelo qual a azatioprina atua sobre processos autoimunes não é claro.
A azatioprina tem múltiplas indicações em reumatologia, alergologia, oncologia etc. Nas doenças intersticiais, a azatioprina pode ser usada em doentes com grave acometimento pulmonar (alveolite e padrão em vidro despolido intenso à tomografia) como adjuvante aos corticoides. Por outro lado, ela pode ser associada aos corticoides em formas não tão graves, como efeito poupador de corticoide, e em doentes com graves efeitos colaterais aos corticoides.
Infelizmente, os estudos são bastante escassos, dificultando uma precisão na recomendação. Alguns autores recomendam usar azatioprina (em vez de metotrexato) para acometimento pulmonar da sarcoidose, doenças autoimunes, entre outras, pelo fato de que o próprio metotrexato possa causar doença pulmonar intersticial, embora isso seja muito raro.
A dose inicial é de 1 mg/ kg de peso/dia. Em geral, inicia-se com 50 mg/dia, podendo chegar a 150 a 250 mg/dia (dose máxima de 2,5 mg/kg/dia). Em baixas doses, recomenda-se usar 1 vez ao dia. Doses acima de 100 mg/dia devem ser divididas em duas doses.
Os efeitos colaterais são relativamente frequentes, embora, facilmente contornados. Os principais são: dispepsia, desconforto abdominal, náusea, vômitos, anorexia, diarreia, fraqueza, milagia e febre.
É importante monitorar periodicamente a série hematológica (pode levar a mielossupressão e pancitopenia) e enzimas hepáticas. Caso seja necessário, a dose pode ser reduzida temporariamente.
Os efeitos adversos mais graves são: imunossupressão com risco de infecções; complicações da pancitopenia (anemia, sangramento); pancreatite aguda; hepatotoxicidade grave; doença hepática veno-oclusiva e malignidade com o uso prolongado.
Imuran®: comprimidos de 50, 75 e 100 mg.
Imuran injetável®: ampolas de 20 mL contendo 100 mg de azatioprina (5 mg/mL).
A azatioprina só deve ser usada em paciente com adequado suporte familiar, aderente e capaz de compreender os seus riscos. Os pacientes devem ser instruídos a procurar assistência médica de urgência caso apresentem alguma infecção ou febre.
Periodicamente, os doentes devem ser monitorados com hemograma e enzimas hepáticas.
Mulheres só devem usar azatioprina se não houver possibilidade de gravidez.
Classe D.
Há múltiplas interações. As principais são:
não permitir vacinação com germes vivos;
cuidado com a associação de azatioprina e inibidores da ECA: risco de toxicidade hematológica e leucopenia grave (mecanismo desconhecido);.
potencializam a imunossupressão e o risco de infecção: micofenolato mofetil, natalizumabe, clofarabina, alefacepte, leflunomida, etarnecepte, inflixicimabe;
aumentam o risco de toxicidade hematológica da azatioprina: metotrexato, clozapina, carboplatina, oxiplatina, cidofovir, cisplatina, quimioterápicos, imatinibe, interferon;
reduz a metabolização da azatioprina: alopurinol.
Classificada entre os agentes alquilantes do tipo mostarda nitrogenada. Sua forma ativada, a mostarda fosforamida, alquila e se liga com várias moléculas estruturais intracelulares incluindo ácidos nucleicos. Sua ação citotóxica se deve principalmente à ligação de cadeias de DNA e RNA, assim como à inibição da síntese de proteínas. É bem absorvida por via oral com biodisponibilidade em torno de 75% e meia-vida de 3 a 12 horas. Sua eliminação é principalmente renal, com 5 a 25% na sua forma inalterada.
Da mesma forma que a azatioprina, há múltiplas indicações da ciclofosfamida já discutidas.
Nas doenças intersticiais, ela deve ser utilizada somente em casos refratários ao tratamento com as demais drogas, pelo seu perfil de toxicidade elevada. Há relatos de seu uso com sucesso em casos de sarcoidose neurológica refratária ao tratamento.
Com relação a PIFI, não há estudos que demonstrem superioridade da ciclofosfamida em relação aos corticoides ou a qualquer outro imunossupressor. A toxicidade relacionada ao uso de ciclofosfamida associada à baixa resposta terapêutica limita bastante o seu uso.
Em especial, a ciclofosfamida tem um importante papel nas doenças reumatológicas sistêmicas (lúpus e dermatopolimiosite) com grave acometimento pulmonar e tomografia mostrando padrão de atividade intensa (alveolite ou padrão em vidro despolido). Da mesma forma, associada aos corticoides, a ciclofosfamida é a droga de eleição na granulomatose de Wegener.
A dose inicial é de 1,5 mg/kg/dia, por via oral ou 500 mg/m2 intravenoso em pulsos inicialmente mensais. A dose é aumentada gradativamente em incrementos de 25 mg até que a leucometria fique entre 4.000 e 7.000/mcL.
O leucograma deve ser acompanhado a cada 2 semanas nos primeiros 3 meses e mensalmente daí em diante. A dose máxima oral habitualmente utilizada é de 150 a 200 mg/dia. O tratamento em forma de pulsoterapia a cada 2 ou 4 semanas foi avaliado em ensaios abertos que mostraram baixa resposta ao uso dessa droga. Nenhum estudo comparou a pulsoterapia com administração oral.
Os seus efeitos colaterais são vários. Os mais graves são: câncer secundário em longo tratamento, mielodisplasia, leucemia, esterilidade, cistite hemorrágica, insuficiência cardíaca, grave imunossupressão, toxicidade medular e pancitopenia.
Outros efeitos adversos são: alopecia, náusea, vômitos, anorexia, diarreia, cefaleia, rash cutâneo, estomatite, escurecimento da pele e unhas.
É importante ressaltar o grande potencial carcinogênico, tendo sido relatados doença linfo e mieloproliferativa e câncer de bexiga.
Todos os indivíduos que forem usar ciclofosfamida devem ser orientados sobre o potencial de infertilidade e deve ser oferecido coleta de óvulos ou espermatozoides para armazenamento.
Enduxan®: comprimidos de 25 e 50 mg.
Enduxan injetável®: ampolas de 100, 200, 500, 1.000 e 2.000 mg.
A ciclofosfamida só deve ser usada em paciente com adequado suporte familiar, social, aderente e capaz de compreender os seus riscos. Os pacientes devem ser instruídos a procurar assistência médica de urgência caso apresentem alguma infecção ou febre.
O leucograma deve ser acompanhado a cada 2 semanas nos primeiros 3 meses e mensalmente daí em diante. Periodicamente, os doentes devem ser monitorados para manifestações orais (ulcerações), gastrintestinais e cardiovascular (medicação cárdio e hepatotóxica). Pode ser necessária a coleta periódica de enzimas hepáticas.
Homens e mulheres devem assinar termo de responsabilidade para uso da medicação, confirmando que foi orientado quanto ao risco de câncer e infertilidade.
É de extrema importância orientar os pacientes sobre o risco de cistite e de câncer de bexiga. Isso facilita a compreensão de que é essencial uma grande ingestão de água para reduzir essa toxicidade. Além disso, exames periódicos de urina devem ser realizados.
Classe D.
Há múltiplas interações. As principais são:
não permitir vacinação com germes vivos;
medicações cardiotóxicas: podem potencializar a ciclofosfamida. Exemplos: doxorrubicina, daunorrubicina;
podem aumentar o risco de toxicidade hematológica: alopurinol, zidovudina (anemia);
potencializam a imunossupressão e o risco de infecção: micofenolato mofetil, natalizumabe, alefacepte, outros quimioterápicos e imunossupressores;
aumenta o risco de grave mucosite: palifermina.
Apenas reduzir a dose com um clearance estimado em menos de 10 mL/min. Nesse caso, a dose deve ser reduzida em 25%. Após a hemodiálise, deve-se prescrever 50% da dose total.
Apresenta como efeitos colaterais principais: úlcera gastrointestinal ou até perfuração intestinal, leucopenia, infecção bacteriana, plaquetopenia, hepatotoxicidade, fibrose pulmonar, entre outros.
A toxicidade do metotrexato pode ser diminuída com o uso de ácido folínico ou fólico. Alguns autores recomendam biópsia hepática após dose cumulativa de 1 a 1,5 g.
O metotrexato é um antimetabólito análogo ao ácido fólico. É específico para a fase S da divisão celular. Atua inibindo de modo irreversível a di-hidrofolato redutase, impedindo assim a síntese de proteínas, RNA e DNA. Células com alta taxa de proliferação são mais afetadas, como as de linhagem hematopoiética, da mucosa do trato gastrintestinal e espermatogônios. Sua meia-vida varia de 3 a 15 horas de acordo com a dose administrada. Sua eliminação é predominantemente renal com 80 a 90% em sua forma inalterada nas primeiras 24 horas.
Da mesma forma que a azatioprina e a ciclofosfamida, há múltiplas indicações do metotrexato já discutidas.
Nas doenças intersticiais, ela deve ser utilizada somente em casos refratários ao tratamento com as demais drogas, como efeito poupador de corticoide ou em doentes com graves efeitos colaterais aos corticoides.
O metotrexato é utilizado por via oral na dose inicial de 7,5 mg 1 vez/semana. Aumentos sucessivos de 5 mg, a cada 1 ou 2 meses, pode ser necessário, podendo-se chegar até 30 mg/semana. As doses sempre devem ser dadas 1 vez/semana. Entretanto, alguns autores recomendam dividir a dose durante o dia (p.ex., pacientes com prescrição de 22,5 mg/semana; eles podem dividir a dose em 3 tomadas de 7,5 mg durante o dia) para reduzir efeitos colaterais.
Alguns pacientes acabam não tolerando a medicação via oral. Nesse caso, a dose pode ser aplicada por via subcutânea ou intramuscular.
Seu emprego é reservado para casos refratários ao tratamento com corticoides ou como droga poupadora de corticoide. A experiência publicada de um grande centro mostrou que houve resposta com seu uso isolado em 33 de 50 pacientes. Em 9 outros pacientes, a combinação com corticoide obteve resposta clínica. Recidivas são comuns após interrupção do tratamento, mas o retratamento com metotrexato geralmente é eficaz.
O metotrexato não deve ser usado em pacientes com insuficiência hepática, alcoolistas, pancitopênicos, grávidas e mulheres amamentando. Muita cautela ao usar metotrexato em pacientes com insuficiência renal.
Os seus efeitos colaterais são vários, embora transitórios e passageiros. Eles incluem: náusea, vômitos, estomatite, fadiga, dor abdominal, rash cutâneo, fotossensibilidade, alopecia, prurido.
Três potenciais toxicidades devem sempre ser lembradas:
hematológica: pode levar a pancitopenia. Deve-se monitorar periodicamente;
hepática: pode levar a fibrose e cirrose hepática. Com a monitoração e a suspensão da droga se houver alteração hepática, o risco é mínimo;
toxicidade pulmonar: o metotrexato pode levar à pneumopatia intersticial.
Os efeitos adversos mais graves são: agranulocitose, grave estomatite, infecções, neurotoxicidade, eritema multiforme, Stevens-Johnson.
É importante lembrar que o uso de ácido fólico (5 mg/dia) reduz significativamente a toxicidade hematológica e os sintomas gastrintestinais, inclusive, diarreia e estomatite.
Metotrexate oral®: comprimidos de 2,5 mg.
Metotrexate injetável ®: para uso não oncológico; as ampolas disponíveis contém 20, 25 e 50 mg.
O metotrexato só deve ser usado em paciente com adequado suporte familiar, social, aderente e capaz de compreender os seus riscos. Os pacientes devem ser instruídos a procurar assistência médica de urgência caso apresentem alguma infecção ou febre.
O hemograma e os testes hepáticos devem ser colhidos a cada 2 meses.
É importante ressaltar que a presença de tosse, febre ou piora da dispneia deve indicar imediata procura ao hospital (risco de pneumonite pelo metotrexato).
Classe X (contraindicado).
Há múltiplas interações. As principais são:
não permitir vacinação com germes vivos;
medicações hepatotóxicas: acitretina, paracetamol, álcool;
medicações mielotóxicas: clozapina, sulfonamidas, zidovudina, ganciclovir, trimetoprim, pirimetamina, dapsona, interferon, lamotrigina;
medicações que aumentam o risco de infecção e imunossupressão: alefacepte, natalizumabe, imunossupressores, cidofovir, ciclosporina, tacrolimo, leflunomida, globulina antitimócito, basiliximabe, daclizumabe, efalizumabe, miofenolato mofetil;
medicações que podem aumentar a concentração sérica de metotrexato: anti-inflamatórios não esteroidais; probenecida, ciprofloxacino, penicilinas, salicilatos (diminuem a excreção renal de metotrexato);
aumento do risco de mucosite: palifermina;
amiodarona: pode aumentar o risco de toxicidade hematológica e aumentar os níveis séricos de metotrexato (mecanismo não conhecido).
As doses devem ser ajustadas de acordo com o clearance de creatinina estimado:
entre 60 a 80 mL/min: reduzir a dose em 25%;
entre 50 a 60 mL/min: reduzir a dose em 33%;
entre 10 a 50 mL/min: reduzir a dose em 50 a 70%;
não usar em doentes com clearance < 10 mL/min.
A colchicina é utilizada com base em sua ação antifibrótica. Estudos retrospectivos e um ensaio randomizado falharam em mostrar diferença entre pacientes tratados com corticoide ou colchicina. Como seu perfil de toxicidade é melhor que o dos esteroides, a colchicina pode ser um tratamento alternativo útil.
A colchicina atua inibindo a divisão celular e a migração de granulócitos. Também interfere com a secreção de colágeno pelos fibroblastos e pode aumentar a degradação do colágeno pela ação da colagenase.
A dose recomendada é de 0,5 mg por via oral 1 a 2 vezes/dia. Os efeitos colaterais geralmente são náusea, vômitos, dor abdominal e diarreia.
A experiência com o uso de penicilamina em FIP é limitada. Um estudo prospectivo não randomizado comparou o uso de prednisona, prednisona/D-penicilamina e prednisona/D-penicilamina/colchicina e não mostrou diferença entre os grupos com relação à sobrevida ou à função pulmonar.
O mecanismo de ação suposto é a inibição da produção de colágeno e a supressão da função de células T.
A dose recomendada é de 125 a 250 mg por via oral em dose única. Após 8 semanas, a dose pode ser aumentada semanalmente até atingir 500 mg/dia. A dose máxima habitual é de 1.000 mg/dia.
Agente primariamente usado como mucolítico, mas com ação antioxidante.
A acetilcisteína possui um radical sulfidrila que possui ação mucolítica. Sua ação antioxidante pelo menos parcialmente parece ser resultado do aumento dos níveis de glutationa, que explica em parte sua ação protetora hepática na intoxicação por acetaminofeno.
prevenção da insuficiência renal induzida pelo contraste;
como antídoto da intoxicação por acetaminofeno;
como mucolítico.
Dose de 600 mg via oral a cada 12 horas no dia do procedimento e nas 24 horas seguintes.
São leves, mas os mais frequentemente relatados são rash, urticária e prurido, em incidência que varia de 0,5 a 20%. Outros efeitos descritos são náuseas, vômitos, sensação de calor, flushing, sudorese, entre outros. O uso inalatório está associado a aparecimento de broncoespasmo, tosse e dispneia.
Como acetilcisteína e fluimicil, em comprimidos de 600 mg. Em envelopes efervescentes de 100 e 200 mg e xarope com 20 mg/mL. Ainda disponível em forma para uso endovenoso a 10% em ampolas com 300 mg em 3 mL.
Sem recomendações específicas.
Classe B.
Sem interações significativas descritas.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.