(Carregando Índice)... (Carregando Índice)... |
Autor:
Lenise Valler
Médica internista do Hospital Nossa Senhora da Conceição. Médica residente do Serviço de Neurologia do HCPA.
Última revisão: 08/08/2014
Comentários de assinantes: 0
Um paciente do sexo masculino, 44 anos, branco, procura atendimento médico por sentir dor de ouvido e dor de cabeça há oito meses. Relata otalgia bilateral, há cerca de oito meses a um ano, associada à cefaleia holocraniana mais intensa na região frontal. Não apresenta outras queixas inicialmente, mas, com os achados de ectoscopia, ao ser interrogado, afirma que seu rosto está aumentando, com crescimento de nariz e orelhas. Ele relata também haver percebido a necessidade de substituir os sapatos por números maiores e um aumento de aproximadamente 5 kg no último ano. Ao realizar exame, constatou-se que o paciente apresenta bom estado geral, hidratação, face com aspecto grosseiro, hipertricose nasal e auricular. A tireoide está difusamente aumentada, e a voz é grave e rouca. A ausculta com ritmo cardíaco está irregular e sem sopros, a frequência cardíaca é de 90 bpm e a pressão arterial de 160/100 mmHg. Não há particularidades no exame abdominal. Mãos e pés apresentam aspecto grosseiro e úmido.
Outras informações sobre o paciente são as seguintes:
Peso: 92,1 kg; altura: 1,78 m; índice de massa corporal (IMC): 29,06 kg/m².
Exames iniciais: glicemia: 103 mg/dL; tiroxina livre (T4L): 1,1 g/dL; hormônio tireoestimulante (TSH): 2,3 UI/L; prolactina (PRL): 9 ng/mL; insulina: 42 U/mL.
Fator de crescimento insulina-símile tipo 1 (IGF-1): 529 ng/mL (o valor de referência para a idade do paciente é de 37 a 390).
A acromegalia resulta da hipersecreção persistente do hormônio de crescimento (GH). O excesso de GH estimula secreção hepática do fator de crescimento insulina-like-I(IGF-I), que causa a maioria das manifestações clínicas da doença.
As características clínicas da acromegalia são atribuídas às altas concentrações tanto de GH quanto de IGF-I.
Os efeitos somáticos incluem a estimulação do crescimento de muitos tecidos, como a pele, o tecido conectivo, as cartilagens, os ossos, as vísceras e os tecidos epiteliais.
Os efeitos metabólicos são a retenção de nitrogênio, o antagonismo da insulina e a lipólise.
Além disso, os adenomas somatotróficos por si só podem causar sintomas locais como cefaleia, defeitos de campo visual (geralmente hemianopsia bitemporal) e paresias cranianas.
Em geral, cerca de 60% dos pacientes eventualmente apresentam cefaleia, e 10%, sintomas visuais.
O início da acromegalia é insidioso, e a progressão da doença é muito lenta. O intervalo entre o início dos sintomas até o diagnóstico é de aproximadamente 12 anos.
A seguir, são apresentados os sintomas da acromegalia:
•Supercrescimento acral. Todos os pacientes com acromegalia têm supercrescimento acral e de tecidos moles embora a extensão do hipercrescimento varie.
As características desse supercrescimento incluem aumento mandibular (macrognatia) e aumento de mãos e pés, que resultam em aumento do tamanho dos sapatos e das luvas e necessidade de ajuste de anéis.
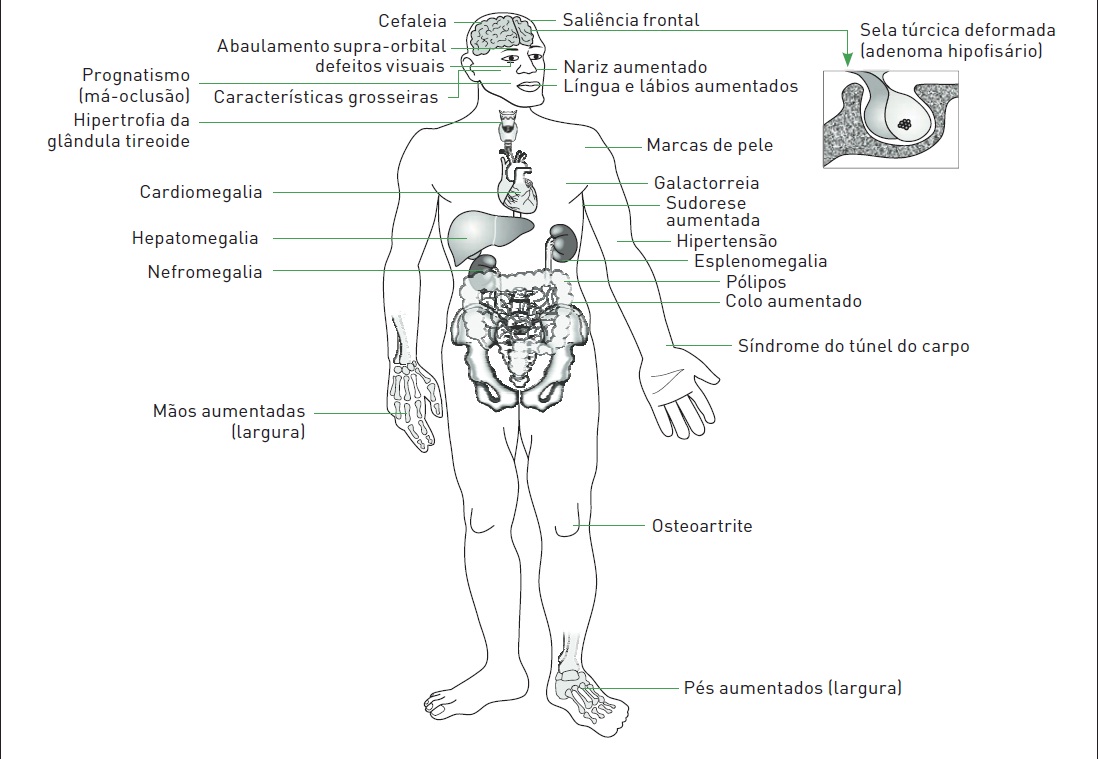
•A face passa a apresentar um aspecto grosseiro, com aumento do nariz e dos ossos frontais, bem como da mandíbula, e os dentes separam-se (Fig. 34.1).
•Supercrescimento articular. Tecido sinovial e cartilagens aumentam, causando artropatia dos joelhos, tornozelos, quadril, vértebras e outras articulações. Sintomas articulares são comuns na apresentação da doença, e lombalgia e cifose são frequentes. Também pode haver dor lombar ocasionada por osteoporose, a qual ocorre por excesso de GH ou insuficiência gonadal devido ao aumento hipofisário.
•Densidade óssea. A densidade óssea pode aumentar nas vértebras e no quadril de mulheres, mas não ocorre se estas apresentam deficiência de estrógeno.
•Efeitos metabólicos. A acromegalia é associada a hiperinsulinismo, resistência à insulina, diabetes, em 10 a 15% dos casos, e tolerância à glicose diminuída, em mais de 50% dos pacientes. Alguns deles têm hipertrigliceridemia ou hipercalciúria.
•Doença cardiovascular. Entre as anormalidades cardiovasculares, estão hipertensão arterial, hipertrofia de ventrículos e miocardiopatia. A manifestação mais comum é a hipertrofia biventricular, que se desenvolve independentemente da hipertensão e se manifesta precocemente durante a doença.
A miocardiopatia é caracterizada por disfunção diastólica e arritmias. Essas alterações ocorrem tanto devido à hipertensão (presente em 43% dos pacientes) quanto à acromegalia em si. A redução da secreção do GH melhora algumas das anormalidades cardíacas. Entre os pacientes com acromegalia, 3 a 10% apresentam insuficiência cardíaca. Um aumento da prevalência de doença valvar cardíaca também tem sido relatado. É esperado que 30% dos pacientes com acromegalia apresentem regurgitação aórtica, e 5%, regurgitação mitral.
•Funções sexuais. Os macroadenomas podem causar diminuição da secreção de outros hormônios hipofisários, mais comumente das gonadotrofinas. Em muitas mulheres com acromegalia, há disfunção menstrual, com ou sem galactorreia.
•A hiperprolactinemia ocorre em 30% dos pacientes.
•Cabelo e pele. Há adelgaçamento da pele e o cabelo se torna quebradiço nesses pacientes. Hiperidrose é de incidência comum. O cabelo dos pacientes cresce mais rapidamente, o crescimento do cabelo aumenta e alguns podem ter hirsutismo.
•Outras manifestações. Podem manifestar-se macroglossia e apneia do sono, agravamento da voz, parestesias das mãos, síndrome do túnel do carpo, neuropatia periférica sensitivo-motora simétrica.
•Neoplasia de colo. Na neoplasia de colo, ocorre aumento do risco de pólipos colônicos e câncer. No caso de ocorrência de outras neoplasias, em adição ao risco de câncer de colo, a acromegalia pode ser associada a outros tumores. Em homens, há aumento de tumores, como adenocarcinoma de estômago, esôfago e melanoma. Também há aumento de leiomiomas uterinos em mulheres.
•Órgãos como tireoide, coração, fígado, rins e próstata podem aumentar (Fig. 34.2).
•Fadiga. Este pode ser um sintoma proeminente. Pode manifestar-se devido ao distúrbio do sono, à disfunção cardiovascular, à neuropatia, ao hipogonadismo, à hiperglicemia ou à combinação desses fatores.
•Hiperfosfatemia. Cerca de 70% dos pacientes apresentam essa alteração.

Figura 34.1
Homem com acromegalia evidenciando mudanças faciais, como aumento mandibular, nasal e dos ossos frontais, além de características grosseiras.
Fonte: Adaptada de Forbes e Jackson.¹
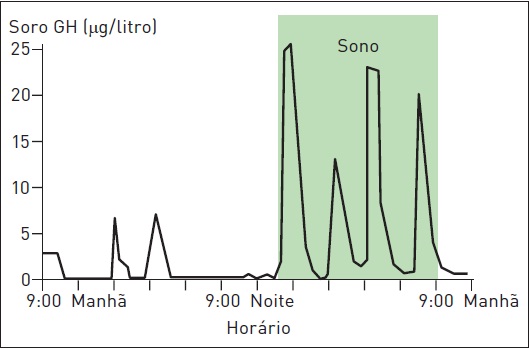
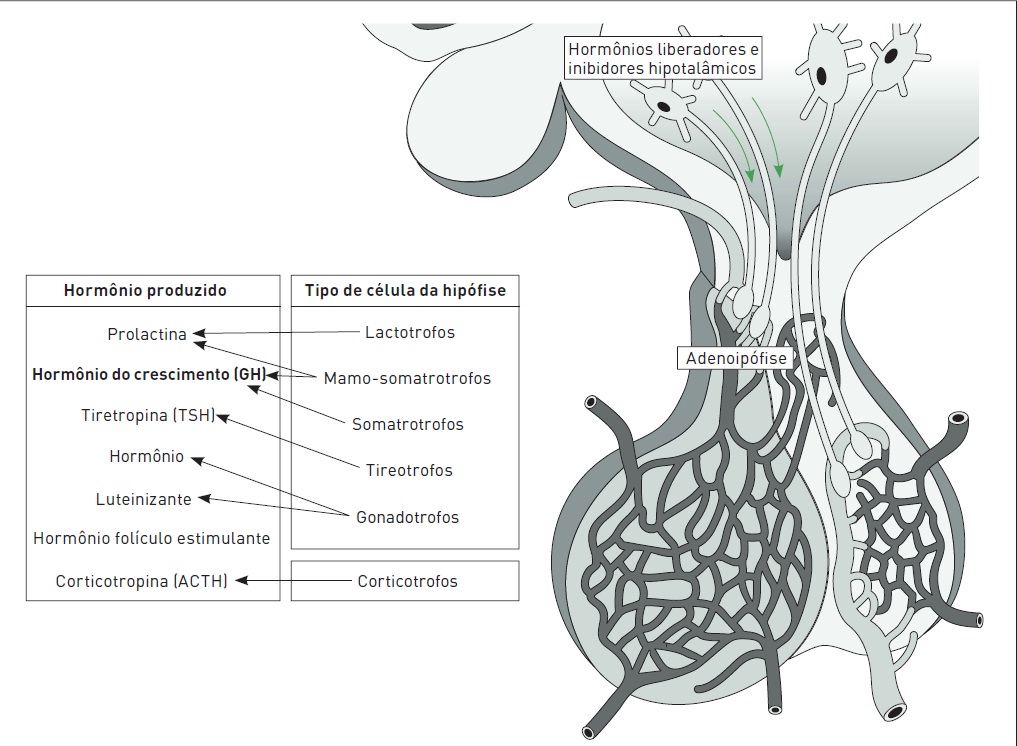
O GH é produzido pela glândula hipófise de forma pulsátil, com níveis maiores durante o sono (Fig. 34.3). Pessoas normais têm níveis baixos na maior parte do dia. Outros fatores responsáveis pelo controle da secreção de GH incluem o hormônio liberador de GH (GHRH), a somatostatina e o fator de crescimento insulina-like I (IGf-I)(Fig. 34.4). Em pacientes com acromegalia, o aumento na frequência dos pulsos resulta em altos níveis de GH.
As síndromes familiares associadas à hipersecreção de GH são neoplasia endócrina múltipla tipo 1 (tumores hipofisários, pancreáticos e de paratireoide), síndrome de McCune-Albright (aparência clínica com pigmentação cutânea e hipersecreção hipofisária) e complexo de Carney (pigmentação cutânea, mixomatose mucocutânea, mixoma cardíaco, lesões de tireoide e mama e adenomas hipofisários secretores de GH) (Fig. 34.4).
A acromegalia familiar isolada é descrita com perda de heterozigosidade no cromossomo 11q13 e recentemente com mutações de baixa penetrância em indivíduos com adenomas hipofisários familiares. Outras causas raras de hipersecreção de GH são tumores pancreáticos e outras lesões centrais (p. ex., hamartoma hipotalâmico, coristoma e ganglioneuroma).
Figura 34.2
Principais efeitos do aumento do hormônio de crescimento (GH).
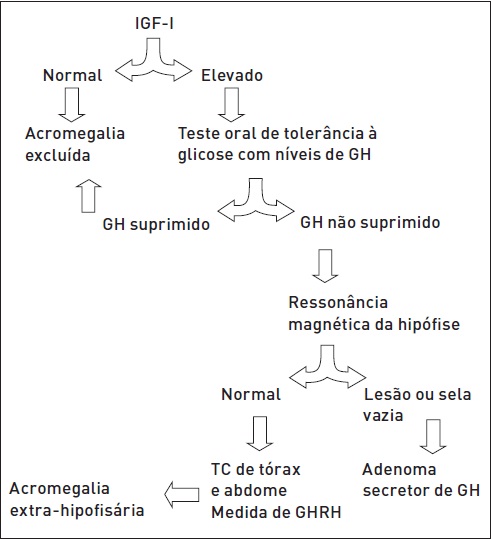
O diagnóstico provável baseia-se nos achados clínicos característicos da doença, sendo a confirmação feita por meio deexameslaboratoriaisedaavaliaçãoradiológica(Fig.34.5).
O diagnóstico laboratorial da acromegalia visa à confirmação da produção excessiva de GH, quer por medidas diretas de seus níveis sanguíneos, quer pela dosagem de fatores circulantes GH-dependentes, em especial pela mensuração dos níveis do IGF-I, também conhecido como somatomedina C.
Figura 34.3
Níveis do hormônio do crescimento GH conforme horário.
Figura 34.4
O controle da secreção de GH é realizado por hormônio liberador de GH (GHRH) e somatostatina, que, através da veia porta, liberam fatores específicos somatotróficos e fator de crescimento insulina-like-I (IGF-I).
As dosagens hormonais têm também um papel muito importante no acompanhamento do tratamento dos pacientes, permitindo avaliar o resultado das diferentes modalidades terapêuticas empregadas na acromegalia.
Tanto a concentração de GH quanto a de IGF-I estão maiores em praticamente todos os pacientes com acromegalia.
O aumento de IGF-I sérico em geral é desproporcionalmente maior do que o aumento do GH por duas razões: a secreção do GH é flutuante, e ele estimula a secreção da proteína ligante de IGF-I-3(IGFBP-3), a maior proteína ligante de IGF-I no sangue.
Uma vez que há suspeita clínica de acromegalia, a próxima etapa a ser seguida é a testagem bioquímica para confirmação do diagnóstico e determinação da causa.
Concentração sérica de IGF-I. Os níveis séricos de IGF-I têm boa relação com os níveis médios de GH secretados durante o dia, estando, portanto, elevados na maioria dos pacientes com acromegalia. No entanto, a relação é logarítmica em vez de linear, observando-se um platô nas concentrações de IGF-I quando os níveis de GH se elevam acima de 15 a 20 g/L. Os níveis aumentados de IGF-I têm alta especificidade no diagnóstico de acromegalia, pois as situações mais frequentes que cursam com elevação doIGF-I, como gestação e puberdade, em geral não oferecem grandes dificuldades no diagnóstico diferencial. Entretanto, é fundamental o ajuste dos valores de acordo com a idade do paciente para a correta interpretação dos resultados laboratoriais, pois há um declínio dos níveis de IGF-I com o envelhecimento.
Ao contrário do GH, os níveis de IGF-I também não variam de hora em hora de acordo com alimentação, exercícios ou sono.
Após terapia, o objetivo deve ser a normalização dos níveis de IGF-I, novamente de acordo com a idade do paciente. Em geral, sugere-se que o IGF-I seja dosado após quatro semanas da cirurgia, mas alguns autores acreditam que a dosagem inicial possa ser realizada mais precocemente, entre 1 a 3 semanas de pós-operatório. No acompanhamento ao paciente, um aspecto relevante é a observação de eventuais discrepâncias entre os níveis de GH e IGF-I, principalmente com GH normal e IGF-I alto ou, menos comumente, GH alto com IGF-I normal.
Figura 34.5
Algoritmo para o diagnóstico de acromegalia.
TC, tomografia computadorizada.
Concentração sérica de GH. Essa concentração é indicada para pacientes com valores duvidosos de IGF-I ou para aqueles com altos níveis de IGF-I sérico em que a confirmação bioquímica se faz necessária.
A concentração sérica de GH varia de menos de 0,5 a 1 ng/mL, durante a maior parte do dia, 2 a 5 ng/mL, antes das refeições ou após exercícios, até 20 ou 30 ng/mL à noite ou após exercício intenso. Níveis séricos de GH podem também ser mais altos em pacientes com diabetes melito não controlado, doenças hepáticas e desnutrição.
Todos os pacientes com acromegalia apresentam secreção de GH aumentada. Entretanto, a média de GH sérico frequentemente varia de 2 a 10 ng/mL durante a maioria dos dias, valores que podem ser encontrados em pessoas normais.
Teste oral de tolerância à glicose (TOTG) com medida do GH. A determinação dos níveis de GH durante o TOTG é o exame referencial na abordagem laboratorial de acromegalia. O teste tem sido realizado dosando-se os níveis de GH, durante 2 ou 3 horas em intervalos de 30 minutos, após administração de 50 a 100 g de glicose, tendo como fundamento a supressão da secreção hipofisária de GH que ocorre após sobrecarga de glicose em indivíduos normais. A resposta normal consiste na supressão dos níveis de GH para valores menores do que 2 g/L (o GH após TOTG é maior do que 2 ng/mL em mais de 85% dos pacientes com acromegalia).
O principal impedimento para a realização desse teste é a presença de diabetes melito associado, por isso recomenda-se a medida da glicemia concomitante.
Na acromegalia, os níveis de GH não são suprimidos e, em alguns casos, podem mesmo apresentar uma elevação paradoxal após a sobrecarga de glicose. Entretanto, os valores de corte no TOTG para estabelecer o diagnóstico da acromegalia e para monitorar a doença após o tratamento têm variado consideravelmente desde a introdução dos radioimunoensaios (RIAs) para GH. Quando dosado por ensaio imunorradiométrico (IRMA) ou imunoquimioluminescente, a concentração de GH cai para menos de 0,3 ng/mL após realização de TOTG em pessoas normais. Então, o diagnóstico de acromegalia, atualmente, pode ser feito com métodos mais sensíveis, que permitem melhor discriminação entre os pacientes com e sem a doença.
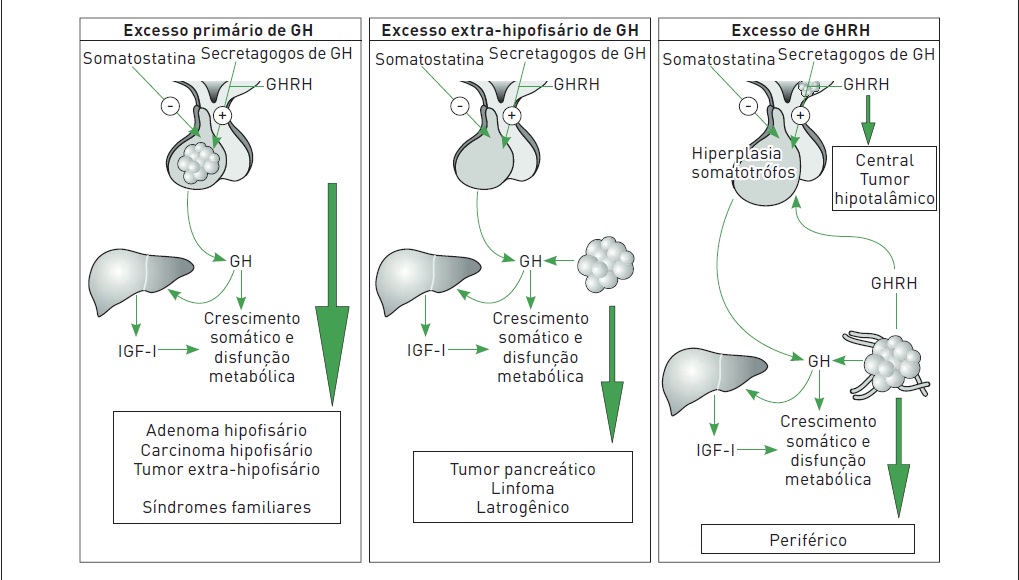
Em mais de 95% dos casos, a causa da hipersecreção do GH é um adenoma somatotrófico da hipófise (Fig. 34.6).
Causas de acromegalia
1.Excesso primário de GH Adenoma de GH
Adenoma misto produtor de GH e prolactina Adenoma mamossomatotrofos
Adenoma pluri-hormonalCarcinoma produtor de GH
Síndromes familiares (neoplasia endócrina múltipla, acromegalia familiar, síndrome de McCune-Albright, síndrome de Carney)
2.Excesso de GH
Tumores em células pancreáticas Linfoma
Iatrogênico
3.Excesso de GHRH
•Produção ectópica central (< 1%) Hamartoma hipotalâmico, coristoma, ganglioneuroma
•Produção ectópica periférica (1%) Carcinoma brônquico, tumores pancreáticos, tumores pulmonares de pequenas células,
carcinoma medular de tireoide, feocromocitoma
Figura 34.6
Causas de acromegalia. Na maioria dos casos, é ocasionada devido à produção excessiva de GH ou GHRH. Em casos raros, a doença é associada a síndromes familiares.
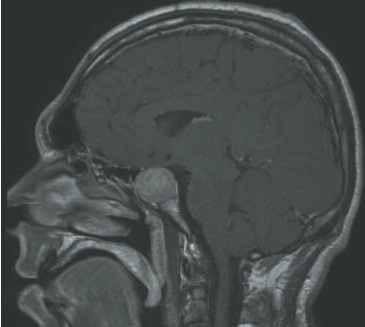
Tumores hipofisários com menos de 2 mm de diâmetro podem ser detectados por ressonância magnética, e suas dimensões e extensão anatômica acuradamente identificadas (Fig. 34.7).
Em 75% dos pacientes com adenomas somatotróficos, o tumor é um macroadenoma (tumor de 10 mm ou mais de diâmetro) e pode haver extensão parasselar ou suprasselar.
A ressonância magnética não possibilita a distinção entre tumores funcionantes e não funcionantes. Essa distinção deve ter como base estudos bioquímicos.
Figura 34.7
Ressonância magnética com lesão hiperintensa na região da hipófise.
A acromegalia atinge igualmente homens e mulheres em todas as faixas etárias, com uma incidência estimada de 3 a 4 casos novos/milhão e uma prevalência estimada de 40 a 70 casos/milhão.
A média de idade no diagnóstico de pacientes afetados é de 40 a 45 anos. Como a doença se desenvolve de forma insidiosa ao longo dos anos, frequentemente há um intervalo de até mais de 10 anos entre o início estimado dos sintomas e o diagnóstico.
A taxa de mortalidade dos pacientes com acromegalia parece estar aumentada. Ocorre morte primariamente por doença cardiovascular e câncer.
Determinantes de sobrevida
•Níveis de GH
•Hipertensão
•Doença cardíaca
•Diabetes melito
•Duração dos sintomas
Causas de morte
•Cardiovasculares – 38 a 62% dos casos
•Respiratórias – 0 a 25% dos casos
•Neoplasias – 9 a 25% dos casos
O tratamento da acromegalia tem como objetivo a diminuição da concentração do IGF-I para um valor de refe- rência, considerando-se a idade e o gênero, e a diminuição da concentração do GH para menos de 1 ng/mL (1 g/L). Com a normalização do IGF-I sérico, a expectativa de vida é similar à da população em geral.
O tratamento também pode aliviar os sintomas. Para isso, tem-se as seguintes opções:
Cirurgia transfenoidal. Essa cirurgia é o tratamento de escolha para adenomas, pequenos ou grandes, mas ainda ressecáveis, e que causam sintomas visuais. Quando a cirurgia é feita em centros experientes, a secreção de GH diminui para o normal em 80 a 90% dos pacientes com microadenomas (tumores < 10 mm).
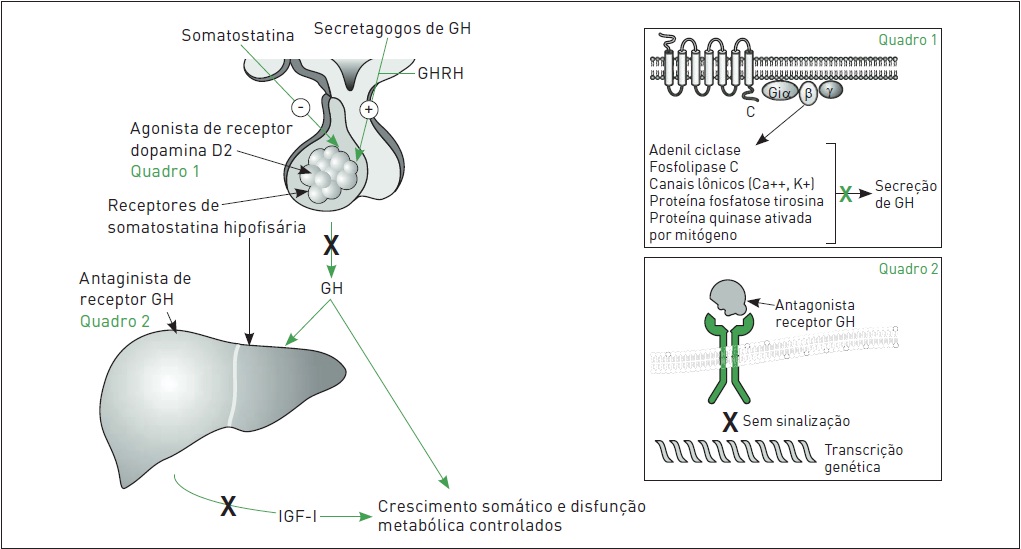
Tratamento medicamentoso. Várias medicações são viáveis para tratar acromegalia, incluindo as que inibem a secreção de GH e as que inibem sua ação (Fig. 34.8). O tratamento farmacológico é utilizado quando a cirurgia isoladamente não reduz o nível sérico de GH e IGF-I ao normal, mas o papel desse tratamento como terapia primária não está esclarecido. Entre os medicamentos disponíveis para o tratamento da acromegalia, estão os seguintes:
•Análogos da somatostatina. Octreotida e lanreotida são análogos da somatostatina que inibem a secreção do GH de forma mais efetiva que a somatostatina nativa devido a potência e tempo de meia-vida plasmática maiores.
•Agonistas de dopamina. Esses fármacos inibem a secreção de GH de forma menos eficiente que os análogos da somatostatina.
• Antagonistas do receptor GH. O pegvisomant é um antagonista dos receptores de GH utilizado em casos em que não há resposta a outros tratamentos (ver Quadro 34.1).
Figura 34.8
Receptores para o tratamento de acromegalia: receptores de somatostatina hipofisária, receptores D2 e receptores de GH periférico são potenciais alvos para tratamento.
1.Agonista dopaminérgico
Eficácia principalmente em tumores com produção de GH e prolactina.
Administração via oral e baixo custo.
Menos efetivo se comparado a outros tratamentos.
2.Análogos da somatostatina
Taxa de sucesso de 45 a 65% dos casos.
Podem ser utilizados como tratamento de primeira escolha.
Administração via parenteral.
3.Antagonista do receptor de GH
Geneticamente análogo da molécula do GH humano. Eficácia em adquirir níveis normais de IGF-I em 97% dos casos.
Disponibilidade precária.
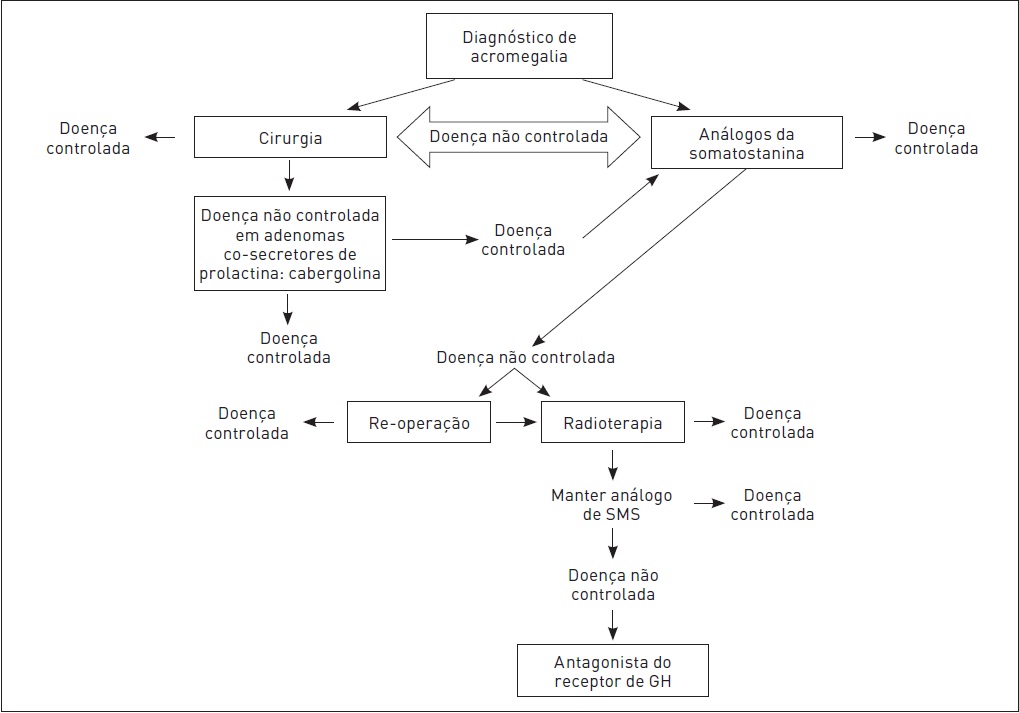
Radioterapia. Esse tratamento pode ser feito em pacientes cuja doença não é controlada por cirurgia ou tratamento medicamentoso (Fig. 34.9).
Figura 34.9
Algoritmo para o manejo da acromegalia.
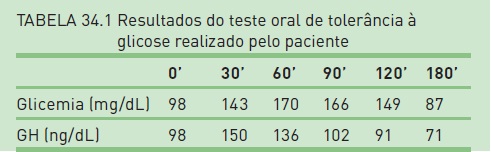
A Tabela 34.1 mostra os resultados do teste oral de tolerância à glicose realizado pelo paciente.
Em relação aos exames de imagem, foi possível verificar os seguintes diagnósticos: na colonoscopia, não houve alterações; a endoscopia digestiva alta evidenciou esofagite e gastrite leves; a ultrassonografia de abdome, hepatoesplenomegalia discreta; o ECG, sobrecarga de câmaras esquerdas; o ecocardiograma, hipertrofia septal e fração de ejeção de 55%; a campimetria visual, hemianopsia temporal bilateral; e a RM de crânio, macroadenoma hipofisário, ausência de compressão de quiasma óptico e haste hipofisária livre.
O paciente foi submetido à cirurgia transesfenoidal para a retirada do tumor. O exame anatomopatológico foi compatível com adenoma de hipófise. A imuno-histoquí-mica foi positiva para GH em mais de 50% das células.
O paciente apresentou boa recuperação pós-operatória, com melhora do controle pressórico e dos demais sintomas.
GH (1ºmês) = 4 ng/mL; IGF-I = 298 ng/mL
GH (2ºmês) = 2,4 ng/mL; IGF-I = 270 ng/mL
GH (5ºmês) = 1,6 ng/mL; IGF-I = 210 ng/mL
GH (8ºmês) = 1,1 ng/mL; IGF-I = 190 ng/mL
1. Forbes J, Jackson WF. Atlas colorido e texto de clínica médica. 2. ed. São Paulo: Manole; 1997.
Cook DM, Ezzat S, Katznelson L, Kleinberg DL, Laws ER Jr, Nippoldt TB, et al. AACE Medical Guidelines for Clinical Practice for the diagnosis and treatment of acromegaly. Endocr Pract. 2004;10(3):213-25
Melmed S. Medical progress: acromegaly. N Engl J Med.2006;355(24):2558-73.
Molitch ME. Clinical manifestations of acromegaly. Endocrinol Metab Clin North Am. 1992;21(3):597-614.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.