(Carregando Índice)... (Carregando Índice)... |
Autores:
Marcelo Basso Gazzana
Médico pneumologista, internista e intensivista. Médico do Serviço de Pneumologia do HCPA e do CTI Adulto do Hospital Moinhos de Vento. Mestre em Ciências Pneumológicas pela UFRGS.
Denise Rossato Silva
Médica pneumologista. Professora adjunta de Pneumologia da Faculdade de Medicina da UFRGS. Doutora em Ciências Pneumológicas
pela UFRGS.
Última revisão: 28/04/2014
Comentários de assinantes: 0
Um paciente do sexo masculino, 71 anos, tabagista (índice tabágico: 65 maços por ano), que exerce a profissão de comerciante, comparece à consulta relatando tosse seca irritativa com início há cerca de oito meses e dispneia progressiva aos médios esforços há quatro meses (escala de dispneia do Modified Medical Research Council [MMRC] na categoria dois). Apresenta hipertensão arterial sistêmica e utiliza hidroclorotiazida na dose de 25 mg ao dia há cerca de três anos. Não há outros achados na história médica pregressa. Afirma não apresentar febre, emagrecimento, anorexia, sudorese, nem sintomas osteoarticulares. Também não há relato atual ou passado de exposição a substâncias orgânicas ou inorgânicas no trabalho ou na sua casa. Ao realizar exame físico, verificam-se eupneia, ausculta pulmonar com crepitantes finos teleinspiratórios em bases, ausculta pulmonar com hiperfonese de P2 e hipocratismo digital. Não são observados sinais de doença reumática. Na radiografia de tórax que o paciente realizou há duas semanas, evidenciou-se infiltrado de padrão intersticial difuso, com predomínio nos lobos inferiores e na região subpleural.
As doenças pulmonares parenquimatosas difusas (DPPDs) representam um grande número de doenças (mais de 150), caracterizadas por infiltração celular e extracelular nas regiões acinares dos pulmões, isto é, distais aos bronquíolos terminais.1 Por definição, doenças de via aérea (p. ex., asma) e da circulação pulmonar (p. ex., hipertensão arterial pulmonar) não são incluídas nessa categoria. Em alguns livros-texto, são tradicionalmente denominadas doenças pulmonares intersticiais, apesar de, em sentido estrito, não ser adequado, pois, na verdade, dentro desse grupo, estão também doenças alveolares difusas (p. ex., hemorragia alveolar, carcinoma bronquioloalveolar, pneumocistose), as quais apresentam pouco ou nenhum envolvimento intersticial. A definição é, portanto, histológica e radiológica.
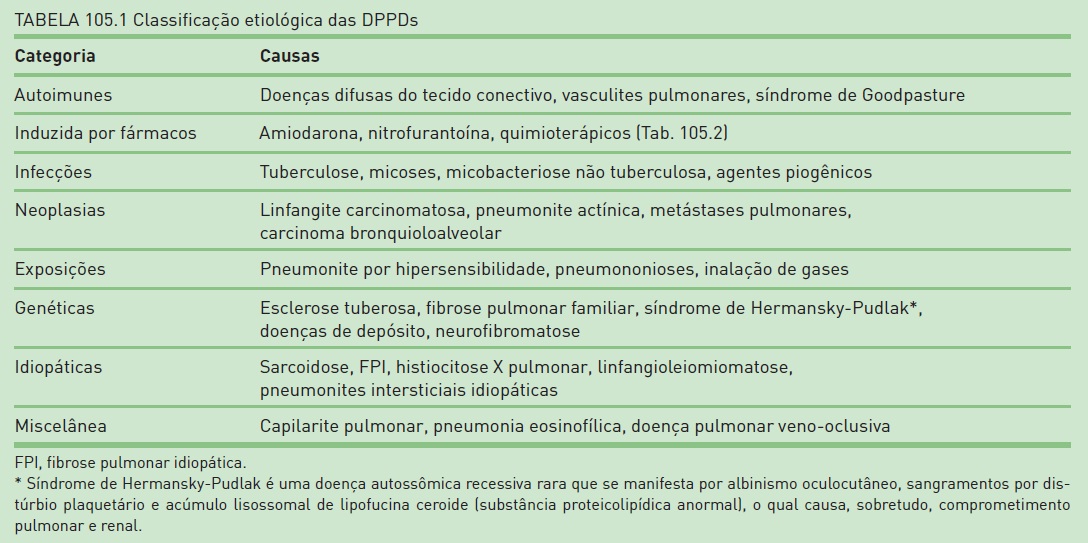
Do ponto de vista etiológico, as DPPDs podem ser classificadas como idiopáticas ou secundárias. Em casos de doenças idiopáticas (p. ex., sarcoidose ou fibrose pulmonar idiopática), não há um mecanismo causal claramente definido, sendo que, em geral, existem diversas hipóteses de etiologia uni ou multifatorial. O grupo das DPPDs secundárias engloba àquelas relacionadas a doenças autoimunes, infecções, neoplasias, exposições (ocupacionais, domiciliares e recreacionais), induzidas por fármacos e de causas genéticas (Tab. 105.1).
Há outros tipos de classificação utilizados na descrição das DPPDs, por exemplo, relacionados a doenças sistêmicas versus doenças primariamente pulmonares, pacientes imunocompetentes versus imunocomprometidos, instalação aguda/subaguda versus crônica, padrões histológicos (pulmão normal, granulomatoso, infiltração linfocítica), padrões radiológicos (localização, padrão do infiltrado, achados associados, normal), função pulmonar (padrão normal, restritivo, obstrutivo ou misto), responsividade aos corticoides (responsivos versus não responsivos) ou uma classificação mista (doenças difusas do tecido conectivo [DDTCs]), granulomatosas, causas inalatórias, causas inerentes, pneumonites intersticiais idiopáticas e algumas entidades específicas).2
A causa mais comum de DPPDs é a fibrose pulmonar idiopática (FPI), também conhecida como alveolite fibrosante criptogênica, representando em média 25 a 35% dos casos. A etiologia de três quartos dos casos de DPPDs envolve FPI, sarcoidose, pneumonite por hipersensibilidade (PH) e DPPD associada a DDTC.

As doenças em geral apresentam um mecanismo etiológico e fisiopatológico próprio. Entretanto, existem algumas características comuns entre eles.
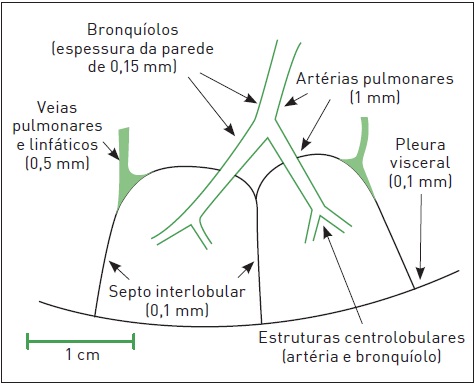
O interstício pulmonar representa o tecido de sustentação dos pulmões. Pode ser dividido didaticamente em três componentes: axial (peribroncovascular), subpleural (periférico) e “verdadeiro” (septal ou alveolar). O interstício “verdadeiro” é o espaço confinado entre a membrana basal e o epitélio alveolar. Essa divisão auxilia o entendimento das alterações radiológicas. Outro conceito importante é o de lóbulo pulmonar secundário, uma estrutura de 1 cm2, composta por bronquíolo e arteríola centrolobulares e septo interlobular com as veias e os vasos linfáticos (Fig. 105.1).3
Figura 105.1
Esquema do lóbulo pulmonar secundário.
Fonte: Adaptada de Raoof e colaboradores.3
As doenças que envolvem o interstício septal podem ser originadas de lesões por via externa (inalatória) ou interna (por meio da corrente sanguínea). Além do interstício, frequentemente outros componentes da parede alveolar estão incluídos no processo, como o epitélio e o endotélio, sendo o termo “alveolite” uma boa definição desse processo.
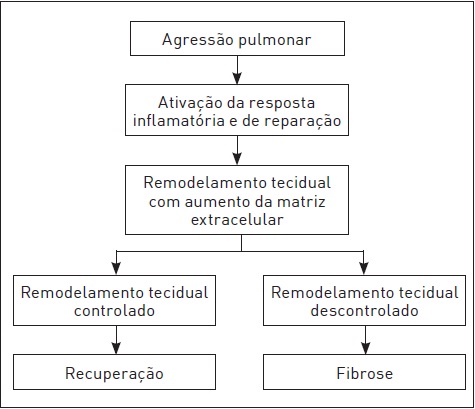
A resposta fisiopatológica do interstício pulmonar a uma agressão é monótona, isto é, independentemente do tipo de estímulo inicial, a sequência dos mecanismos é muito semelhante (Fig. 105.2). O agente inicial pode ser microrganismo, parasita, célula neoplásica, material exógeno inalado, fármaco, radiação, entre outros. Depois de uma lesão inicial no epitélio alveolar e/ou no endotélio capilar, há uma fase de inflamação alveolar (alveolite) relacionada aos macrófagos e aos linfócitos. Após, segue a fase de reparação, a qual pode levar à recuperação completa do parênquima ou à fibrose, que é causada por proliferação de fibroblastos e deposição de colágeno de forma desordenada. A lesão final e o consequente comprometimento funcional pulmonar variam conforme a intensidade e a duração do estímulo agressor e as características do paciente em relação à suscetibilidade e ao grau de resposta à agressão.4
Embora a resposta inflamatória à agressão seja um mecanismo de defesa do organismo, impedindo a progressão da lesão e o reparo adequado do parênquima pulmonar, um descontrole desse processo inflamatório e imunológico pode causar destruição da membrana alveolar. As células envolvidas, tanto na resposta adequada quanto na inadequada, parecem ser as mesmas e, após ativadas, geram um ciclo vicioso de perpetuação desse processo. Nessas duas fases (inflamação e reparação), há grande interação entre células epiteliais, inflamatórias e fibroblastos. O processo inicia com destruição das células da parede alveolar (pneumócitos tipo 1) e do endotélio, desencadeando um aumento da permeabilidade e consequente edema e acúmulo de células inflamatórias no septo e na luz alveolares. A ativação dessas células, sobretudo dos macrófagos (mas também plaquetas, células endoteliais e linfócitos), ocasiona, por meio da liberação de mediadores (como os fatores de crescimento transformadores – TGF-beta), o recrutamento celular sistêmico (pela corrente sanguínea) e local, tanto de outras células inflamatórias quanto das mesenquimais, essas últimas decisivas na patogênese da fibrose. As células mesenquimas (representadas pelos fibroblastos) induzem a proliferação de células semelhantes e produzem colágeno, principalmente na isoforma 1. Essa deposição desordenada de células e de colágeno causa colapso alveolar. O organismo apresenta mecanismos de defesa contra a produção excessiva de colágeno, como a liberação de prostaglandina E2 e do interferongama, que, em geral, está diminuída nos indivíduos doentes.5,6
Sob perspectiva histológica, essas alterações são caracterizadas por uma alveolite inicial, em que há edema, células inflamatórias (linfócitos e macrófagos ativados) e pneumócitos tipo 2 descamados na luz, mas ainda com integridade da parede alveolar. Com a progressão do processo, ocorrem edema e infiltração de células inflamatórias também no septo alveolar e formação de membranas hialinas (que representam a necrose de células endoteliais e sobretudo dos pneumócitos tipo 1). Paralelamente, há proliferação de pneumócitos tipo 2 reativos a fim de tentar evitar o desnudamento alveolar. O processo final é o colapso alveolar, relacionado ao espessamento dos septos pela deposição celular de colágeno e da incorporação de exsudatos da luz alveolar. Na fase final desse processo, ocorre intensa fibrose, caracterizada por paredes alveolares espessadas com abundantes fibroblastos e miócitos derivados dos bronquíolos, mas poucas células inflamatórias. Observam-se frequentemente áreas císticas (denominadas faveolamento por assemelharem-se a favos de mel), que ocorrem devido à hiperdistensão de áreas mais preservadas recobertas por epitélio bronquiolar metaplásico. Nos pacientes com fibrose pulmonar idiopática, cujo padrão histológico é denominado pneumonite intersticial usual, esse processo patológico caracteristicamente é multifocal, alternando áreas de reparação anormal com deposição de colágeno e focos fibroblásticos e áreas de pulmão normal, embora estas sejam cada vez menos frequentes com a progressão da doença.7
Figura 105.2
Patogênese geral das doenças pulmonares parenquimatosas difusas.
O quadro clínico varia conforme a etiologia específica. Em geral, os sintomas mais frequentes são dispneia (inicialmente ao esforço e, após, no respouso) e tosse (predominantemente seca). Na história médica dos pacientes com DPPD, é fundamental definir o contexto deles, relacionando-o ao estado de imunossupressão e ao tempo de estabelecimento dos sintomas. Os pacientes considerados imunodeprimidos são os que apresentam infecção por HIV, neoplasias hematológicas, neutropenia de qualquer etiologia, submetidos à quimio ou à radioterapia, a transplantes de órgãos (sólidos ou de medula óssea) e ao uso de corticoides sistêmicos e/ou imunossupressores. Nesse subgrupo, é necessário que seja descartada a possibilidade de causa infecciosa de imediato, uma vez que esta é a etiologia mais comum e pode desenvolver-se rapidamente. Ressalta-se que o tipo de imunossupressão predispõe a causas específicas de DDTC, infecciosas ou não.8
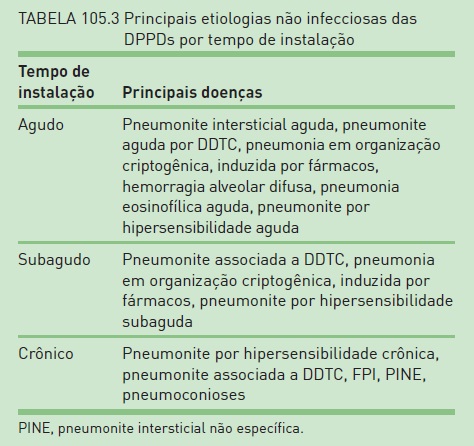
Da mesma forma, o estabelecimento do tempo de início dos sintomas é importante na avaliação dos pacientes. A etiologia infecciosa predomina sobretudo nos quadros agudos (até quatro semanas de sintomas), mesmo em pacientes imunocompetentes (Tab. 105.3).
Os dados obtidos por meio da anamnese fornecem pistas para o estabelecimento do diagnóstico. Nos pacientes com menos de 50 anos, predominam as DPPDs por DDTC e a sarcoidose e, nos mais idosos, a FPI. Tendo em vista o tipo de profissão relacionada à ocorrência de DPPD, as pneumoconioses são mais comuns em homens. Também a paracoccidioidomicose ocorre mais em homens e provavelmente está relacionada a aspectos hormonais (nas mulheres, apresenta-se após a menopausa). A linfangioleiomiomatose ocorre exclusivamente em mulheres. A maioria das DDTCs predomina em mulheres. A história familiar positiva é identificada em alguns casos de FPI e sarcoidose. Já as doenças relacionadas à exposição a antígenos domiciliares também podem apresentar um padrão familiar, mas não por mecanismos genéticos. O tabagismo é associado a várias DPPDs, tais como bronquiolite respiratória/pneumonite intersticial descamativa (PID), pneumonia descamativa, histiocitose pulmonar de células de Langerhans, FPI e um subgrupo de pacientes com pneumonia eosinofílica aguda. Entretanto, a PH ocorre menos comumente em fumantes, embora, quando se manifeste, seja mais grave.9
Os sintomas relacionados a doenças sistêmicas e/ou extrapulmonares devem ser questionados. A ocorrência de febre favorece etiologia infecciosa e/ou de DDTC. A história de câncer aumenta a probabilidade de a DPPD estar relacionada à neoplasia, seja pela existência de metástases, síndrome paraneoplásica, comorbidade ou por efeito do tratamento. Artralgias e outros sintomas reumáticos (rigidez articular matinal prolongada, edema articular, fenômeno de Raynaud, fotossensibilidade, rash facial, xeroftalmia, xerostomia, úlceras orais/genitais, engrossamento da pele, ulcerações nos dedos, fraqueza muscular, alopecia) devem ser diretamente questionados, já que podem apresentar-se de forma sutil e estar associados a colagenoses ocultas. Os sintomas oftalmológicos, além das DDTCs, podem ocorrer em casos de sarcoidose. Os sintomas de refluxo gastresofágico sugerem esclerose sistêmica, doença mista do tecido conectivo ou lesão pulmonar decorrente de aspiração, como pneumonia em organização (geralmente combinada com a existência de células gigantes multinucleadas, bronquiolite ou broncopneumonia e/ou granulomas supurativos e restos alimentares) ou fibrose pulmonar de distribuição broncocêntrica.
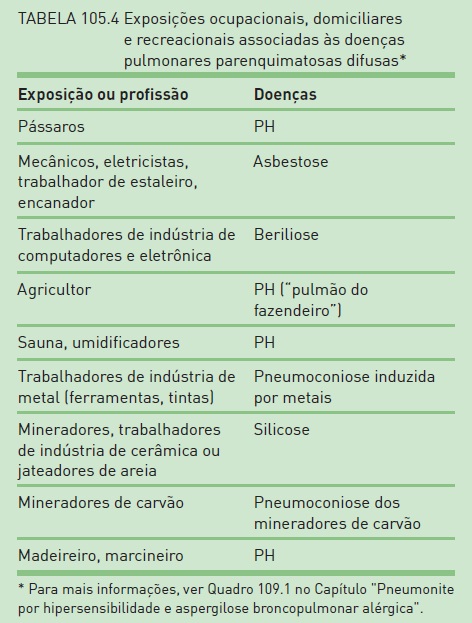
A exposição ambiental ou ocupacional é frequentemente subestimada pelos médicos. Várias DPPDs estão associadas a esse contexto. É essencial realizar uma anamnese detalhada, incluindo exposições domiciliares, recreacionais e ocupacionais. Também é importante a história, não somente o relato da última profissão, já que pode haver um tempo longo de latência entre a exposição e a doença.10 O questionamento sobre colegas de trabalho que adoeceram pode também sugerir o diagnóstico. Nem sempre a exposição é óbvia, e, se o paciente não for questionado, não a relatará espontaneamente. Também se deve considerar a omissão do paciente em relação à exposição, por motivos profissionais ou mesmo por constrangimento pessoal (vergonha de relatar más condições de moradia, medo de que o médico restrinja atividades – p. ex., criação de galinhas). As pneumoconioses (decorrentes da inalação de poeiras inorgânicas) que ocorrem com mais frequência no Brasil são a silicose e a asbestose.11 A silicose origina-se mais comumente de exposição em indústrias cerâmicas, de abrasivos, construção de estradas e túneis, jateamento de areia, corte e moagem de pedras, pedreiras, fundições e escavação de poços. A asbestose (fibrose pulmonar por exposição a fibras de asbesto) geralmente é causada por trabalho em indústria de fibrocimento, fabricação de telhas e caixas d’água, fricção, como embreagem, lonas e pastilhas de freio, entre outras atividades, como mineração. A pneumonia de hipersensibilidade, no Brasil, em geral decorre de exposição a pássaros ou mofo no ambiente doméstico ou no trabalho e exposição a isocianatos, como em indústria de tintas (Tab. 105.4).12
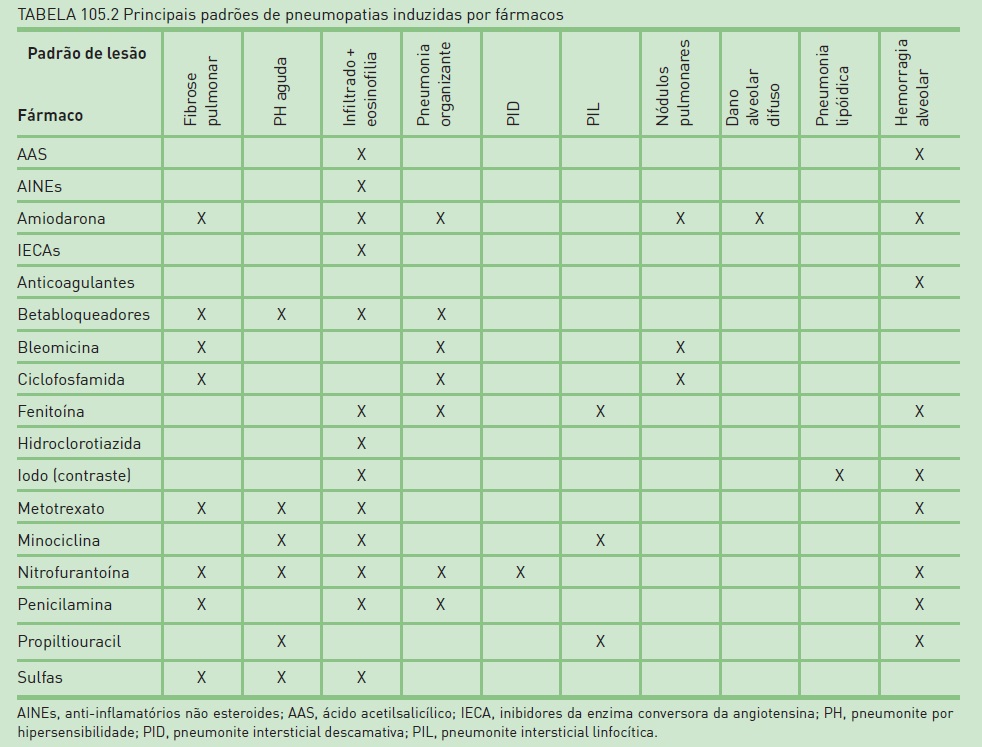
Vários medicamentos podem estar associados às DPPDs. Entretanto, o quadro não é específico, podendo a mesma medicação apresentar diversos padrões radiológicos e histológicos de doença, bem como o mesmo padrão estar relacionado a medicamentos diferentes. Embora, na maioria dos casos, o envolvimento pulmonar ocorra nas primeiras semanas do uso, há relatos de pneumopatia após anos de utilização do fármaco. Então, é necessário obter a história medicamentosa completa, sobretudo dos pacientes que mais apresentam toxicidade pulmonar. Em alguns contextos, há potencialização do efeito tóxico, como a realização concomitante de radioterapia ou o uso de altas frações inspiradas de oxigênio.
Uma pesquisa rápida sobre as principais causas medicamentosas das DPPDs pode ser realizada no endereço eletrônico do Hospital Universitário de Dijon, França,* o qual permite a busca pelo medicamento ou pelo padrão radiológico.
Os fármacos que mais comumente resultam em DPPDs são a nitrofurantoína, a amiodarona, o metotrexato e a bleomicina (Tab. 105.2).
No exame físico, devem-se observar as alterações associadas ao comprometimento pulmonar e os achados sistêmicos relacionados à doença de base. Na ausculta pulmonar, crepitantes finos teleinspiratórios, denominados em velcro, ouvidos mais na região das bases pulmonares, são característicos de muitas doenças intersticiais, como a FPI. Entretanto, em casos de doenças granulomatosas, como a sarcoidose e a paracoccidioidomicose, há certa dissociação clínica e radiológica, sendo os crepitantes em velcro auscultados em menos de 20% dos pacientes.1 O grasnido (squeake) é um som inspiratório, agudo, tipo “piado de gaivota”, que ocorre em pacientes com doenças em que há comprometimento bronquiolar, como na PH. Podem-se auscultar sibilos expiratórios em casos de bronquiolite constritiva, e em geral eles são refratários ao uso de broncodilatadores. Observa-se hipocratismo digital (baqueteamento) em 30 a 50% dos pacientes com FPI e em 50% dos pacientes com pneumonite intersticial descamativa, sendo raro na sarcoidose e nas associadas a DDTC (exceto na artrite reumatoide, em que ocorre frequentemente).2 Deve-se considerar que o hipocratismo está associado a carcinoma brônquico, o qual pode ocorrer paralelamente a uma DPPD, sobretudo aquelas relacionadas ao tabagismo. Os sinais físicos de doença avançada são taquipneia de padrão superficial pela doença restritiva e achados sugestivos de cor pulmonale (insuficiência cardíaca direita), tais como distensão venosa jugular, hepatomegalia, refluxo hepatojugular, edema de membros inferiores, hiperfonese de P2 e sopro de regurgitação tricúspide.13
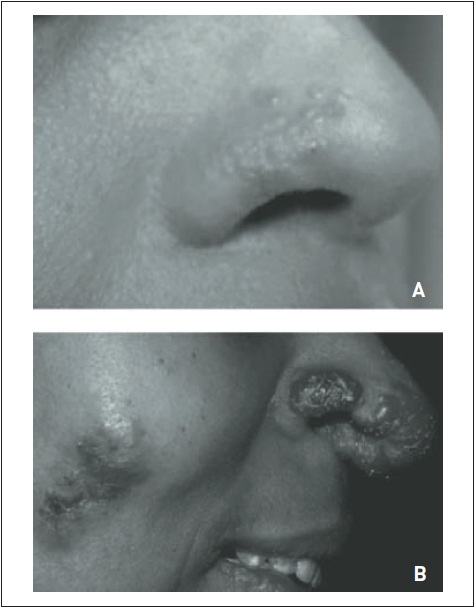
Alguns achados sistêmicos podem auxiliar a estabelecer o diagnóstico. Sinais de artrite em atividade ou sequela de processo prévio (p. ex., desvio articular ulnar na artrite reumatoide) são os mais chamativos. Outras alterações também sugerem DDTC, tais como rash malar, fotossensibilidade, úlceras orais e genitais, fenômeno de Raynaud, sinal de Gottron, heliótropo e sinais de miopatia. Alterações oftalmológicas, como uveíte e conjuntivite, ocorrem em casos de sarcoidose e DDTCs. Podem-se observar linfadenomegalia periférica e hepatoesplenomegalia em pacientes com sarcoidose e neoplasia maligna avançada ou naqueles com granulomatoses infecciosas disseminadas (tuberculose, histoplasmose). Há aumento das glândulas salivares quase exclusivamente em casos de sarcoidose e síndrome de Sjögren. Sinais de doença neurológica, particularmente comprometimento dos nervos cranianos, sugerem neurossarcoidose, embora também possam ocorrer de forma secundária à metástase cerebral ou síndrome paraneoplásica concomitantemente a linfangite carcinomatosa. Pode-se verificar eritema nodoso (paniculite focal) em indivíduos com sarcoidose, DPPD associada a fármacos, doenças granulomatosas, infecções piogênicas e linfomas. O lúpus pérnio é um achado cutâneo muito sugestivo de sarcoidose, caracterizado por lesões púrpuras endurecidas observadas mais comumente no nariz, nas orelhas, nas bochechas e nas mãos (Fig. 105.3).
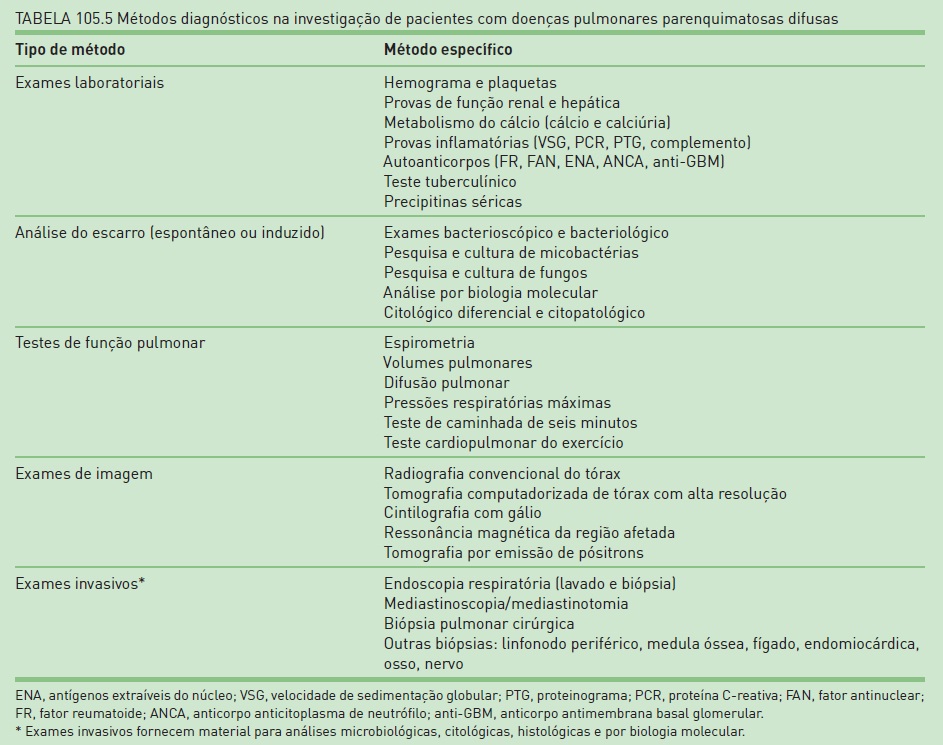
Como os sintomas de DPPDs são muito inespecíficos, a sugestão do diagnóstico normalmente é realizada pela existência de infiltrado pulmonar na radiografia convencional de tórax. Pode-se estabelecer o diagnóstico, na maioria dos casos, por meio da análise minuciosa da história clínica e dos exames de imagem. Exames laboratoriais, testes de função pulmonar e métodos in vasivos para obtenção de material para microbiologia e citoanatomopatologia complementam o rol de testes para avaliação desses pacientes (Tab. 105.5).
Figura 105.3
Lúpus pérnio em pacientes com sarcoidose. (A) Lesão nasal discreta; (B) lesões extensas com destruição nasal.
Os exames laboratoriais podem ser úteis na investigação de uma causa específica já suspeitada (p. ex., fator reumatoide na suspeita de artrite reumatoide) ou na triagem de casos com padrão clínico pouco expressivo. Os exames de rastreamento geralmente não proporcionam um diagnóstico específico, mas podem dar pistas diagnósticas ou reforçar a suspeita. Em geral são solicitados hemograma, contagem de plaquetas, hemossedimentação, proteína C-reativa, eletrólitos séricos (incluindo cálcio), provas de função hepática, provas de função renal e exame qualitativo de urina. A realização de exames laboratoriais gerais também é importante para o acompanhamento da toxicidade pelo tratamento, sobretudo com imunossupressores. A gasometria arterial é essencial para avaliar a necessidade de oxigenoterapia.14
A anemia pode ocorrer em diversas doenças, sejam agudas (pneumonia, hemorragia alveolar) ou crônicas (tuberculose, linfangite carcinomatosa, DDTC), de diferentes padrões (ferropriva, tipo doença crônica ou hemolítica) e de gravidade variável (leve a intensa). Pode-se evidenciar leucopenia em casos de DDTC (sobretudo LES), sarcoidose, tuberculose disseminada e efeito de drogas. A eosinofilia é um achado não tão comum e, portanto, estreita o diagnóstico diferencial. Pode ocorrer em pacientes com pneumonia eosinofílica, sarcoidose, toxicidade pulmonar por fármacos, vasculites (principalmente síndrome de Churg-Strauss), aspergilose broncopulmonar alérgica e parasitoses (síndrome de Loeffler).
Podem-se observar alterações em provas de função renal em casos de doenças do colágeno e síndromes pulmão-rim (síndrome de Goodpasture, granulomatose de Wegener). Pode haver provas de função hepática alteradas na sarcoidose (hepática ou disseminada, sobretudo aumento da fosfatase alcalina), malignidade metastática, tuberculose miliar e DDTC.
Utiliza-se a gasometria arterial para avaliação da troca gasosa e da necessidade de oxigenoterapia. Deve-se solicitá-la para pacientes com oximetria de pulso menor ou igual a 93%, quando há quadro agudo com esforço respiratório, para aqueles com cor pulmonale clínico ou por exames complementares (eletrocardiograma, ecocardiograma) e se houver suspeita de distúrbio respiratório do sono associado à DPPD. Na maioria dos casos, quando alterada, a gasometria arterial evidencia hipoxemia, hipocapnia e aumento do gradiente alveoloarterial.
Provas inflamatórias, como proteína C-reativa, hemossedimentação, proteinograma, que indicam hipergamaglobulinemia policlonal são achados inespecíficos e auxiliam mais durante o acompanhamento do que no diagnóstico. No entanto, o achado de hipogamaglobulinemia pode sugerir a ocorrência de imunodeficiência, seja primária (imunodeficiência comum variável) ou adquirida (infecção pelo HIV), podendo estar associada à pneumonite interesticial linfocítica. Os níveis de complemento medem a atividade inflamatória e também são utilizados para seguimento dos pacientes em doenças específicas. A redução do seu nível sérico (geralmente são dosados os componentes C3, C4 e CH50) indica aumento da degradação, como na agudização de doenças autoimunes ou algumas infecções (endocardite bacteriana, sepse por gram-negativos ou pneumococo). Contudo, pode haver aumento (normalmente menor do que duas vezes os valores de referência) dos componentes do complemento na fase aguda de diversas doenças, tanto em doenças autoimunes (LES, AR e polimiosite) quanto em outras doenças com ativação dessa via (sarcoidose, amiloidose, pneumonia pneumocócica, neoplasias, hepatites, infarto do miocárcio, diabetes). A desidrogenase láctica (DHL ou LDH) pode apresentar aumento em diversas doenças pulmonares (principalmente sua fração 3), tais como pneumonias, infarto pulmonar por embolia, sarcoidose e pneumocistose. Nessa última, há correlação com a gravidade da doença, e os valores maiores do que 220 UI/L apresentam sensibilidade superior a 90% para o seu diagnóstico.14 As enzimas musculares (as mais específicas, aldolase e creatinafosfoquinase total ou fração MM, ou as menos específicas, LDH e TGO) podem estar aumentadas em casos de polimiosite.
O teste tuberculínico (purified protein derivative [PPD], reação de Mantoux) indica o contato com o bacilo da tuberculose e eventualmente apresenta valores menores do que 15 mm de induração em pacientes com micobacteriose não tuberculosa e em indivíduos vacinados com BCG. Caracteristicamente, pode ser negativo em casos de sarcoidose (“roubo” por desvio de linfomonócitos relacionado à doença), tuberculose grave (sobretudo miliar), algumas viroses ou vacinas com vírus vivo, malignidades (incluindo quadros de linfangite carcinomatosa), infecção pelo HIV, desnutrição, uso de imunossupressores e idade avançada. Pode ser útil no diagnóstico diferencial entre sarcoidose, linfomas e linfadenite tuberculosa, mas deve ser interpretado no contexto global, e não isoladamente.
Distúrbios do metabolismo do cálcio ocorrem em pacientes com sarcoidose. O defeito primário envolve o excesso da hidroxilação da 25-hidroxivitamina D para 1,25-hidroxivitaminaD pelas células do granuloma. Isso causa aumento da absorção intestinal de cálcio, provocando hipercalcemia e hipercalciúria. A hipercalcemia é observada em 5% dos pacientes com sarcoidose, e a hipercalciúria, em 10%.15 Ambas as dosagens são recomendadas no acompanhamento dos pacientes. A dosagem de 1,25 hidroxivitamina sérica pode estar elevada nesses casos, bem como no hiperparatireoidismo, outras granulomatoses crônicas (tuberculose, micoses, beriliose) e linfomas. Também se pode verificar a hipercalcemia em casos de linfangite carcinomatosa, relacionada à síndrome paraneoplásica (secreção de PTHrp, nos linfomas e nos carcinomas brônquicos), metástases ósseas concomitantes ao envolvimento pulmonar (sítios mais comuns são mama, primário de pulmão e ovário) e tumores com atividade osteoclástica aumentada (mielomas). Alguns fármacos associados a pneumopatias também podem elevar a calcemia, como tiazídicos, lítio e tamoxifeno.
Devem-se considerar as dosagens do fator antinuclear (FAN) e do fator reumatoide (FR) quando houver suspeita clínica de DDTC ou no contexto das pneumonites intersticiais idiopáticas, uma vez que, em alguns casos, pode haver uma DDTC oculta. O FAN apresenta diferentes percentuais em pacientes com DDTCs, tais como lúpus eritematoso sistêmico (LES) (> 95%), esclerose sistêmica (ES) (80 a 90%), artrite reumatoide (AR) (50 a 60%), síndrome de Sjögren (Ssj) primária (> 90%), polimiosite (PM) (30 a 50%), bem como naqueles com outras condições, por exemplo, infecções crônicas (10 a 50%), neoplasias (20 a 30%), familiares de primeiro grau de pacientes com DDTC (25 a 50%) e indivíduos saudáveis (5 a 13%, apresentando maior positividade com o avançar da idade, mas geralmente com títulos inferiores a 1/160). Pacientes com FPI podem evidenciar FAN (< 1:160) e FR em títulos baixos em até 20%.14,16
O padrão do FAN realizado em células Hep-2 está associado a determinados autoanticorpos e também pode indiretamente sugerir a etiologia, apesar de não definir a especificidade desse autoanticorpo. São exemplos o padrão centromérico (ES, doença limitada), o homogêneo em placa cromossômica metafísica corada (LES idiopático ou induzido por drogas), o homogêneo em placa não corada (ES e outras doenças quando títulos são menores do que 1:640), o pontilhado grosseiro em placa não corada (LES, DMTC), o pontilhado fino em placa corada (ES). Os padrões homogêneo periférico e pontilhado fino, ambos em placa não corada, e citoplasmático ocorrem em diversas doenças autoimunes. A existência de anticorpos específicos, denominados anticorpos contra antígenos extraíveis do núcleo (ENA), pode sugerir uma doença em particular: no LES (DNA nativo, Sm), U1-RNP (DMTC, mas também em LES, ES e AR), U3-RNP (ES, mas também DMTC, LES, AR, Ssj), SS-A/Ro (SSj, mas também em LES, PM, ES), SS-B/La (SSj, mas também em LES), histona (LES e em mais de 95% do LES induzido por drogas), Th/to (ES), Jô-1 (PM), Scl-70 (ES), centrômero (ES, sobretudo doença limitada), RNA polimerase (ES), Ku (sobreposição entre PM e ES) e citrulina (AR).
O FR sugere a ocorrência de AR. Entretanto, pode existir, mesmo em títulos elevados, em outras colagenoses (LES, ES, PM, Ssj, DMTC, vasculites) e em diversas outras doenças (hepatites virais, Aids, influenza, pós-vacinação, neoplasias, principalmente após radio e quimioterapia, tuberculose, entre outras). Os anticorpos anticitoplasma de neutrófilos (ANCA) ocorrem principalmente em casos de vasculites sistêmicas, sendo que o padrão c-ANCA (sugerindo a existência de anticorpos antiproteinase 3) está presente em 80 a 90% dos casos de granulomatose de Wegener, e o padrão p-ANCA (anticorpos antimieloperoxidase), nos casos de poliangeíte microscópica, glomerulonefrite rapidamente progressiva, síndrome de Churg-Strauss ou vasculite induzida por drogas. Os anticorpos antifosfolípideos (especificamente o anticoagulante lúpico, anticardiolipina e 2-glicoproteína-1) estão relacionados à síndrome antifosfolipídeo primária ou secundária a outras DDTCs (LES, ES).
Observam-se os anticorpos antimembrana basal glomerular (anti-GBM) em pacientes com síndrome de Goodpasture e eventualmente naqueles com glomerulonefrite crescêntica e após transplante renal. Quando medido por imunofluorescência indireta, apresenta sensibilidade de 80 a 90% para a síndrome e, quando por Elisa, em mais de 95%. Pode-se utilizá-lo para o acompanhamento terapêutico.
A enzima conversora da angiotensina (ECA) é produzida pelas células epitelioides do granuloma sarcoide. Sua dosagem, no passado, foi utilizada para monitoração dos pacientes com sarcoidose, uma vez que apresenta níveis elevados (maior do que 35 UI/L em adultos) em 40 a 80% dos pacientes com doença em atividade e ocorre redução com a melhora clínica.17 Entretanto, corticoides sistêmicos suprimem sua atividade; logo, um aumento do nível sérico pode estar relacionado à redução do corticoide, e não à exacerbação da doença. Pode também apresentar níveis elevados em 11% dos pacientes com doença inativa e naqueles com outras doenças, como silicose, beriliose, amiloidose, tuberculose, insuficiência renal crônica, diabetes, hipertireoidismo, PH, pneumonite intersticial linfocítica e síndrome do desconforto respiratório agudo (SDRA). Portanto, não é específica para estabelecer a ocorrência de sarcoidose e não deve ser utilizada para tomar decisões terapêuticas.
A pesquisa de precipitinas séricas para antígenos de mofos e de pássaros não está disponível no Brasil, mesmo em laboratórios de referência. Essa pesquisa é solicitada quando há história de exposição sugestiva, por meio da dosagem de um painel de precipitinas dos antígenos mais comuns. A sensibilidade do teste é elevada, mas um resultado negativo não descarta o diagnóstico. Além disso, um resultado positivo deve ser interpretado no contexto clínico, pois pode significar somente sensibilização ao antígeno, mas não necessariamente a ocorrência da doença.18
A dosagem de peptídeos natriuréticos, como o peptídeo natriurético tipo B (BN) ou o N-terminal do proBNP, pode ser útil para pacientes com dispneia aguda e infiltrado pulmonar para possibilitar diferenciação entre causas cardíacas e pulmonares. Nos casos de DPPD crônica, pode auxiliar a detecção e a estratificação de risco quando há hipertensão pulmonar associada à doença de base.
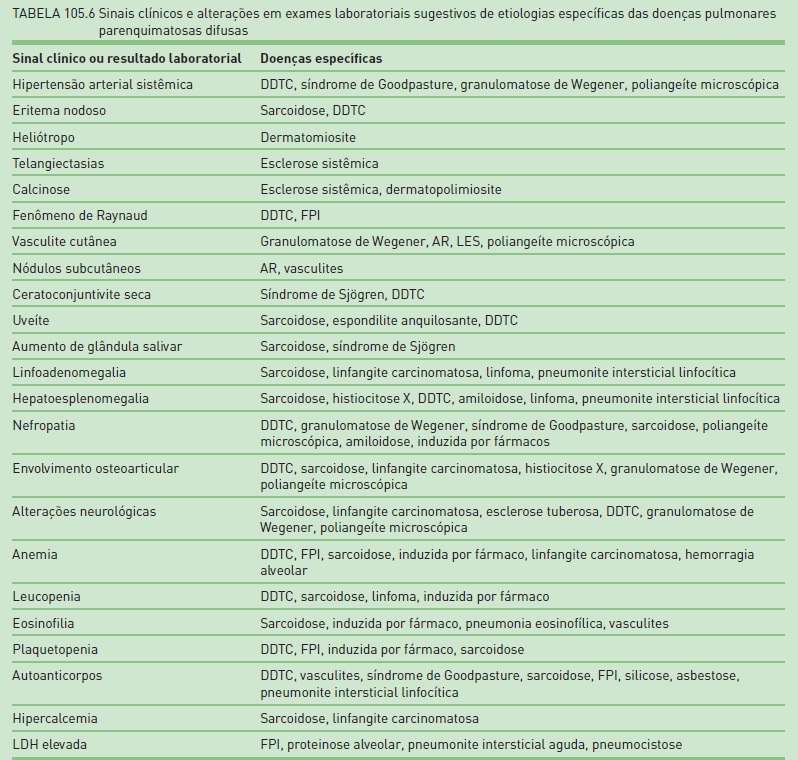
Na Tabela 105.6, são apresentadas as principais alterações no exame físico e em exames laboratoriais que podem sugerir uma etiologia específica das DPPDs.
Análises microbiológicas para bactérias, micobactérias e fungos no escarro podem ser suficientes para estabelecer o diagnóstico quando analisadas juntamente com os quadros clínico e radiológico. A citologia do escarro, observando o perfil inflamatório e a existência de células malignas e de inclusões virais, também pode auxiliar ou confirmar o diagnóstico. A indução do escarro por meio de solução salina hipertônica tem sido um bom método para o diagnóstico de tuberculose quando o escarro espontâneo apresenta resultado negativo ou não há material. Análises por biologia molecular (para mycobacterium tuberculosie e outros agentes) podem ser mais sensíveis e proporcionar um resultado mais rápido do que a cultura, embora possa haver problema em relação à contaminação.
Os testes de função pulmonar são pouco significativos para o diagnóstico, sendo, entretanto, importantes para o acompanhamento do paciente e da resposta à terapia. Em várias DPPDs, também apresentam relação com o prognóstico.
O padrão funcional típico das DPPDs é a restrição, do ponto de vista mecânico, e a dificuldade de realizar trocas gasosas. A espirometria evidencia redução proporcional da capacidade vital forçada e do volume expiratório forçado no primeiro segundo (consequentemente com índice de Tiffeneau e fluxos expiratórios normais ou elevados), confirmados pela medida dos volumes pulmonares, a qual apresenta diminuição da capacidade pulmonar total (CPT). Em geral, o padrão é de restrição intraparenquimatosa quando há redução proporcional da CPT e do volume residual (VR), uma vez que é causada por retração do parênquima pulmonar e redução de todos os seus volumes. As doenças como a polimiosite e o LES, que causam restrição extraparenquimatosa por fraqueza diafragmática, apresentam VR percentualmente menos reduzido (ou de forma ocasional entre normal e elevado) do que a CPT.4
A dificuldade de troca gasosa é a manifestação funcional mais precoce que ocorre nos pacientes com DPPD. Do ponto de vista clínico, é identificada por meio da medida da capacidade de difusão pulmonar do monóxido de carbono (DCO), a qual está reduzida antes dos sintomas e das alterações na espirometria. A dessaturação durante
o exercício também indica dificuldade de troca gasosa. Esta pode ser identificada no teste de caminhada de seis minutos, que, além de avaliar a oxigenação, permite quantificar a capacidade física do paciente pela medida da distância percorrida. Utiliza-se o teste cardiopulmonar do exercício (ergoespirometria) geralmente para avaliar a dispneia inexplicada, podendo sugerir a necessidade de investigar DPPD não suspeita, e também para monitorar a evolução do paciente.19
Alguns achados obtidos por meio das provas de função pulmonar podem sugerir um subgrupo de etiologias específicas. O aumento dos valores da Dco pode ser um indício de hemorragia alveolar. Já a existência de uma DPPD confirmada por exames radiológicos junto com as provas de mecânica pulmonar (espirometria e volumes) com resultado normal ou padrão obstrutivo ocorre em poucas doenças, tais como pneumonite por hipersensibilidade, sarcoidose (sobretudo nos estágios I e II), pneumonia em organização criptogênica, doenças císticas (linfangioleiomiomatose, histiocitose X pulmonar), doenças predominantemente alveolares (pneumocistose, hemorragia alveolar, proteinose alveolar), bronquiolite constritiva, doença intersticial associada à bronquiolite respiratória e combinação de fibrose e enfisema pulmonar no mesmo paciente.
849
A investigação complementar inicia-se geralmente pela radiografia de tórax (necessária para o diagnóstico sindrômico), que evidencia infiltrado em vários lobos, homogeneamente ou não, de padrão intersticial e/ou alveolar. Atualmente, a maioria dos pacientes é submetida à tomografia computadorizada de tórax com técnica de alta resolução (TCAR) para complementar a investigação, pois esse exame é mais sensível do que o raio X convencional, possibilita estabelecer alguns diagnósticos que apresentam padrão típico, estreita o diagnóstico diferencial, tem implicações prognósticas e auxilia na escolha do local para a realização de eventual biópsia pulmonar.20 É essencial que o clínico tenha um bom treinamento para a interpretação da radiologia torácica, sobretudo se, na sua prática diária, atender pacientes com doença respiratória, não se limitando apenas a ler o laudo do radiologista, mas sim correlacionando essas informações com sua impressão clínica e radiológica. Cerca de 10% dos pacientes com DPPD podem apresentar o resultado do raio X de tórax normal e, mais remotamente, também o da TC.21 Algumas técnicas complementares na TC podem ser úteis, tais como injeção de contraste (melhor visualização dos linfonodos e das massas mediastinais), cortes em inspiração e expiração (observação de alçaponamento aéreo) e obtenção das imagens em decúbito dorsal e ventral (descarta a possibilidade de infiltrados gravitacionais sem significado clínico). A causa mais comum dessa situação é a PH. Então, uma radiografia normal não possibilita que o médico exclua o diagnóstico de DPPD se há um contexto apropriado. É fundamental comparar o exame radiológico atual com anteriores se estes estiverem disponíveis.
Inicialmente, no raio X de tórax, é necessário diferenciar o predomínio de componente alveolar (nódulos de 7 a 12 mm, limites mal definidos, coalescem precocemente, com broncograma aéreo, volume pulmonar mantido) de componente intersticial (nódulos menores do que 3 a 6 mm, limites definidos, não coalescem ou o fazem tardiamente, simétricos e sem broncograma aéreo, redução volumétrica do pulmão, áreas de faveolamento, linhas de Kerley). Na TCAR, o infiltrado intersticial pode ser melhor caracterizado como padrão nodular (este subdividido em nódulos perilinfáticos, randômicos e centrolobulares), padrão septal (i. é., espessamento dos septos interlobulares), padrão reticular (espessamento dos septos intralobulares) e padrão cístico. Alguns sinais adicionais também são úteis para sugerir diagnósticos diferenciais, tais como vidro fosco (opacidade que ainda permite a visualização dos vasos no seu interior), consolidação (opacidade que apaga a imagem dos vasos no seu interior), perfusão em mosaico (áreas de vidro fosco alternadas com áreas hipo ou normodensas, que sugerem doença bronquiolar ou da circulação pulmonar), pavimentação em mosaico (áreas em vidro fosco com septos interlobulares espessados), nódulos centrolobulares com árvore em brotamento (sinal de envolvimento da via aérea distal), bandas parenquimatosas (opacidades lineares em direção à pleura), linha subpleural, bronquiectasias/bronquiolectasias de tração, sinal do halo (consolidação circundada por vidro fosco), placa pleural e faveolamento (cistos empilhados na região subpleural, que representa estágio terminal de uma DPPD).
A cintilografia com gálio pode ser útil para o diagnóstico de sarcoidose quando o paciente apresenta lesões extrapulmonares e raio X de tórax normal ou inconclusivo. O padrão pandalambda sugere o diagnóstico. Embora tenha sido muito utilizada no passado para acompanhamento dos pacientes com sarcoidose, a cintilografia com gálio não apresenta associação com a evolução da doença e com a resposta terapêutica. O uso da tomografia por emissão de pósitron com fluordesoxiglicose F18 tem sido estudado para diferenciar a FPI de outras doenças intersticiais, mas os resultados são inicipientes. Com as técnicas atuais, a ressonância magnética é pouco significativa para a avaliação da maioria dos pacientes com DPPD.22
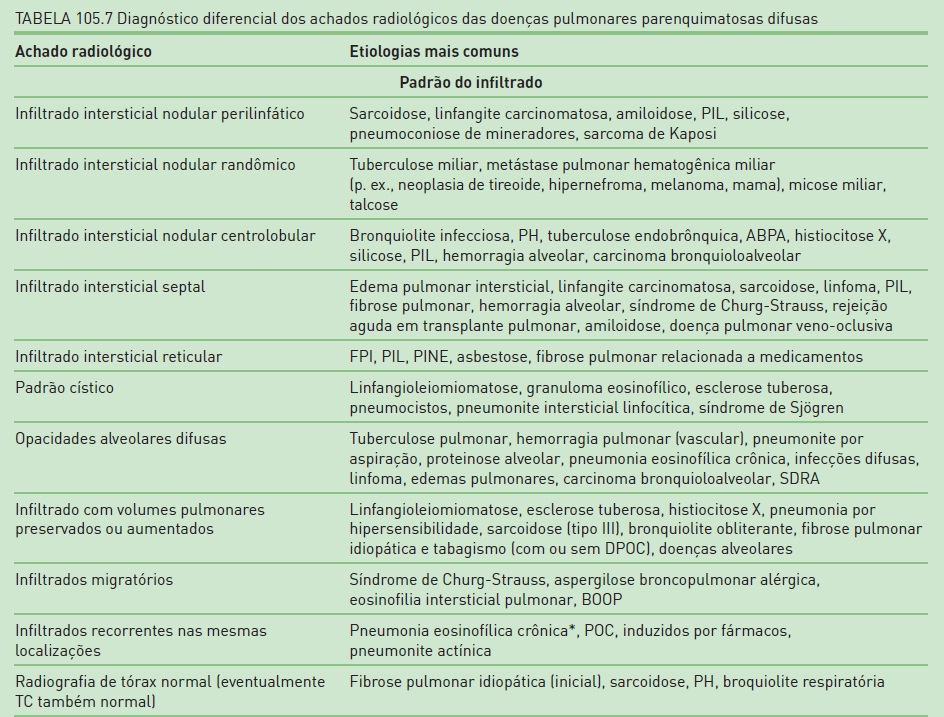
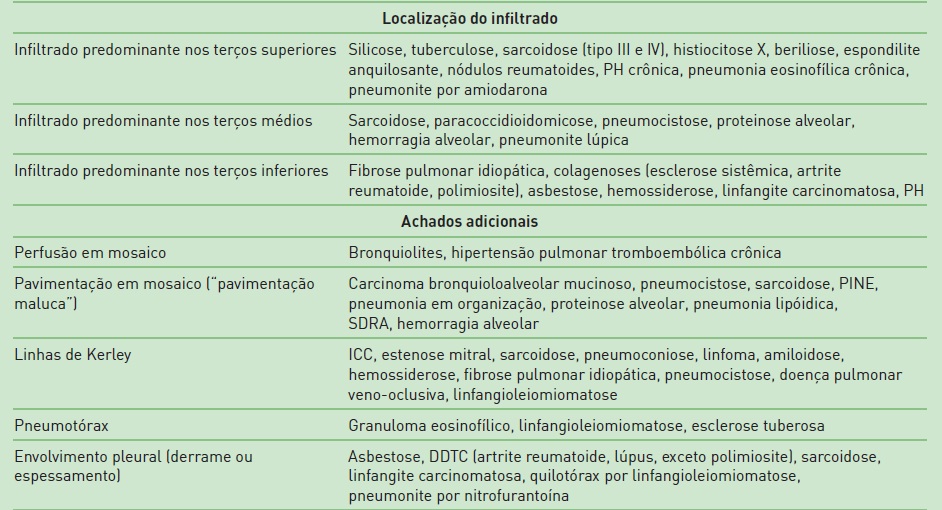
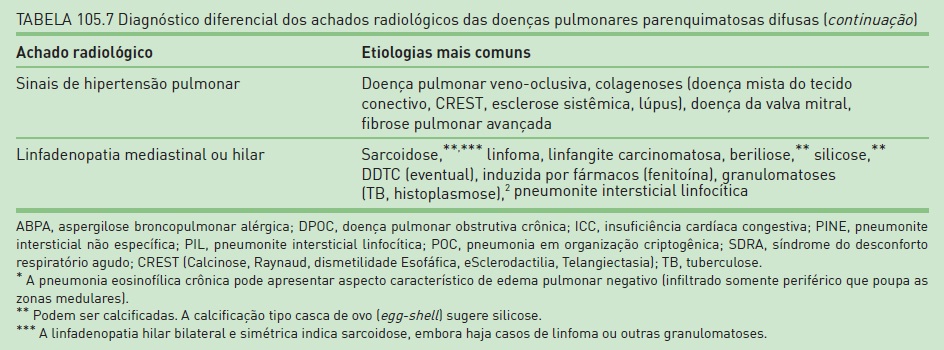
Na Tabela 105.7, constam os principais achados radiológicos das DPPDs.20,21,23
Quando não se estabelecer o diagnóstico por meio de exame clínico, padrão radiológico e exames laboratoriais não invasivos, recomenda-se a realização de endoscopia respiratória com lavado broncoalveolar (LBA), com ou sem biópsia. Esse exame é realizado sob sedação e associado a baixas taxas de complicação quando o preparo é adequado.
O LBA, realizado com técnica padronizada (instilação de 100 a 200 mL de soro fisiológico pelo broncoscópico flexível e aspiração de, pelo menos, 40% desse volume), é útil para estabelecer diagnóstico específico ou mesmo para evidenciar um padrão citológico que restringe as possíveis causas. A identificação de germes específicos, por exemplo, histoplasma, confirma o diagnóstico. Entretanto, em algumas situações, é obrigatória a correlação clínica, já que pode haver colonização, mas não necessariamente infec ção, tais como nos casos em que são observados Aspergillus, bactérias piogênicas ou BAAR (que, além da tuberculose, podem ocorrer nas micobacterioses não tuberculosas ou de forma eventual na nocardiose). A existência de células malignas no LBA configura envolvimento do espaço aéreo pela neoplasia, tipicamente no carcinoma bronquioloalveolar, embora possa, de modo não tão frequente, ser observada na linfangite carcinomatosa em fase avançada.
O lavado hemorrágico em sucessivas alíquotas indica hemorragia alveolar difusa (idiopática, DDTC – LES, infecção por leptospirose, citomegalovirose, neoplasia – carcinoma bronquioloalveolar, congestão crônica – estenose mitral, ICC grave), sobretudo quando associado à pesquisa de macrófagos com hemossiderina positiva (mais de 20%). Se possível, deve-se quantificar essa pesquisa com o índice de Gold, que é positivo quando há envolvimento de mais de 150 células por campo. A pesquisa de cristais (realizada com luz polarizada) pode indicar a exposição a metais pesados (sílica, asbesto), mas não necessariamente doença (pneumoconiose). Líquido floculento, associado a material amorfo positivo à coloração Ácido periódico Schiff (do inglês, periodic acid Schiff [PAS] ou glóbulos amorfos basofílicos na coloração de Giemsa, sugerem proteinose alveolar, mas também podem ocorrer em casos de pneumocistose. Uma coloração específica que evidencia substância lipóidica pode indicar pneumonia lipóidica, que ocorre nos processos aspirativos pulmonares recorrentes.
O LBA normal é composto quase totalmente por macrófagos alveolares. Entretanto, em várias DPPDs, a citologia é alterada. Pode haver eosinocitose, observada em casos de reações a fármacos, parasitoses e pneumonias eosinofílicas. Uma contagem de eosinófilos maior do que 50% indica fortemente pneumonia eosinofílica. Já uma contagem de neutrófilos maior do que 50% (sobretudo maior do que 65%) sugere acentuadamente dano alveolar difuso ou infecção supurativa. Neutrocitose, em nível menor, é verificada na maioria dos pacientes com FPI, sendo fator prognóstico, mas não indicando futura resposta à terapia. Linfocitose (contagens maiores do que 25%), denominada às vezes de alveolite, ocorre em casos de doenças granulomatosas (sarcoidose, tuberculose, PH, beriliose), DDTC, reação a fármacos, infecção viral e pneumonite não específica em fase celular. Quando as contagens de linfócitos são maiores do que 50%, geralmente a causa é a PH ou, mais raramente, a pneumonite interesticial linfocítica (primária ou associada à colagenose). A existência de plasmócitos pode sugerir PH (sobretudo associado a macrófagos vacuolados), pneumonia porLegionella ou Pneumocystis jiroveci, linfoma não Hodgkin ou pneumonia eosinofílica crônica. O aumento de mastócitos no LBA pode ocorrer em pacientes com tuberculose, linfomas, FPI, asma e, menos frequentemente, sarcoidose.14,24
A imunofenotipagem do LBA pode diferenciar linfócitos CD4 de CD8, sendo a relação maior do que 3,5 sugestiva de sarcoidose, e a menor do que 1, de PH. A dosagem repetida de linfócitos CD4 no LBA foi utilizada, no passado, para monitoração dos pacientes com sarcoidose, mas apresenta muita variabilidade e não é mais recomendada pela maioria dos centros. A imunofenotipagem pode também identificar células CD4 positivas, isto é, histiócitos, que são células normais, mas que, em pacientes com suspeita clínica de granuloma eosinofílico (também denominado histiocitose X pulmonar, histiocitose de células de Langerhans), quando em percentual maior do que 3%, sugerem fortemente o diagnóstico (ocorre em mais de 50% dos pacientes com essa doença). Os histiócitos também são positivos para o marcador S100.
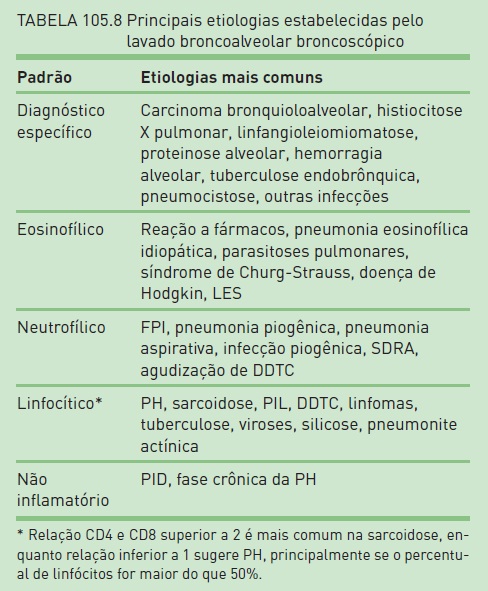
Na Tabela 105.8, há os principais achados obtidos no lavado broncoalveolar broncoscópico.
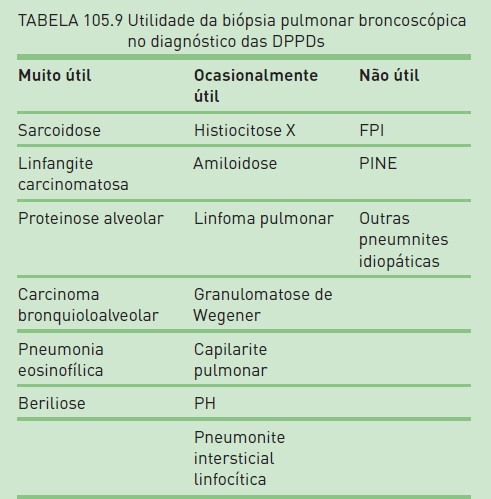
Nos casos de doenças com padrão bronquiolocêntrico, como a sarcoidose, recomenda-se também a realização concomitante ao LBA de biópsia pulmonar broncoscópica (transbrônquica) (Tab. 105.9). Devem-se obter quatro a seis fragmentos (eventualmente até dez) que incluam parênquima pulmonar. A complicação mais temida é o pneumotórax, uma vez que pacientes com DPPD mais extensa podem apresentar dificuldade para expandir novamente os pulmões mesmo com drenagem torácica, tendo em vista a complacência pulmonar reduzida. Os fragmentos são colocados em formol para que seja realizada análise histo lógica e eventualmente algumas peças em soro fisiológico para avaliação microbiológica e lâminas de imprint para citologia. Nos pacientes com DPPD não bronquiolocêntrica (p. ex., FPI), ou nos casos inconclusivos, a biópsia, quando necessária, deve ser realizada cirurgicamente, desde que não haja contraindicações (Quadros 105.1 e 105.2). O ideal é que esse procedimento seja feito por videocirurgia (VATS – videotoracoscopia), embora biópsia “a céu aberto” por minitoracotomia também seja um procedimento seguro. Devem ser obtidos três a quatro fragmentos de segmentos diferentes com pelo menos 3 cm de profundidade, evitando áreas com faveolamento já estabelecidos. A mortalidade associada à biópsia pulmonar cirúrgica é inferior a 1%, e a morbidade, menor que 3%.9,25
A biópsia pulmonar transtorácia é um método pouco utilizado na investigação de DPPD, mas pode ser útil na de lesões com componente periférico (p. ex., POC).
Realiza-se comumente mediastinoscopia para o diagnóstico da sarcoidose quando há envolvimento dos linfonodos mediastinais, permitindo acessar as cadeias paratraqueais e pré-carinais. Indica-se, eventualmente, a mediastinotomia esquerda (procedimento de Chamberlain) para obtenção de material na estação pré-vasculare/ou aortopulmonar. Caso haja linfadenopatia periférica, hepatomegalia, envolvimento neural ou de medula óssea, a biópsia desses locais pode estabelecer o diagnóstico de uma doença sistêmica com envolvimento pulmonar (sarcoidose, tuberculose miliar, histoplasmose, neoplasia metastática). Para escolher o local que a biópsia será realizada, devem-se considerar a possibilidade de diagnóstico e o fato de o procedimento ser invasivo.
Não havendo infecção e neoplasia, os clínicos reconhecem as DPPDs como uma síndrome com as seguintes características: dispneia ao esforço; infiltrado pulmonar difuso no raio X de tórax; alterações fisiológicas, incluindo restrição pulmonar, redução da capacidade de difusão e hipoxemia em repouso ou ao esforço; vários fatores histológicos de inflamação e fibrose do parênquima.1 Divide-se a investigação dessa síndrome no estabelecimento do diagnóstico etiológico e na avaliação da gravidade do comprometimento atual.
Fundamenta-se a investigação da causa da DPPD em uma anamnese minuciosa, estabelecendo-se inicialmente o contexto clínico apropriado (doença aguda vs. crônica, pacientes imunodeprimidos vs. imunocompetentes). Uma revisão criteriosa de exposições (domésticas, ocupacionais e recreacionais), hábito tabágico, achados relacionados à DDTC e a outras doenças sistêmicas (p. ex., granulomatoses infecciosas e neoplasias) e uso de fármacos revelam a maioria das causas das DPPDs. Resultados de exames laboratoriais podem aumentar a probabilidade de algumas doenças, como hipercalciúria na sarcoidose e positividade para autoanticorpos nas DDTCs. Adicionalmente ao raio X de tórax, realiza-se a TCAR na maioria dos pacientes, e sua análise detalhada, estabelecendo padrões de comprometimento e procura de achados específicos, é essencial para o prosseguimento da investigação. É frequente a necessidade de obtenção de material biológico para o diagnóstico, em geral por endoscopia, pelo LBA, e, em alguns casos, de biópsia broncoscópica. Alguns desses pacientes necessitarão de biópsia pulmonar cirúrgica (ou biópsia de outro sítio comprometido).
Deve-se ressaltar que, em pacientes com DDTC, pneumoconioses ou pneumopatias induzidas por fármacos que apresentam quadro clínico e tomográfico característicos, não é necessário executar biópsia (seja endoscópica ou cirúrgica), a menos que haja suspeita de outra doença ou condição associada.
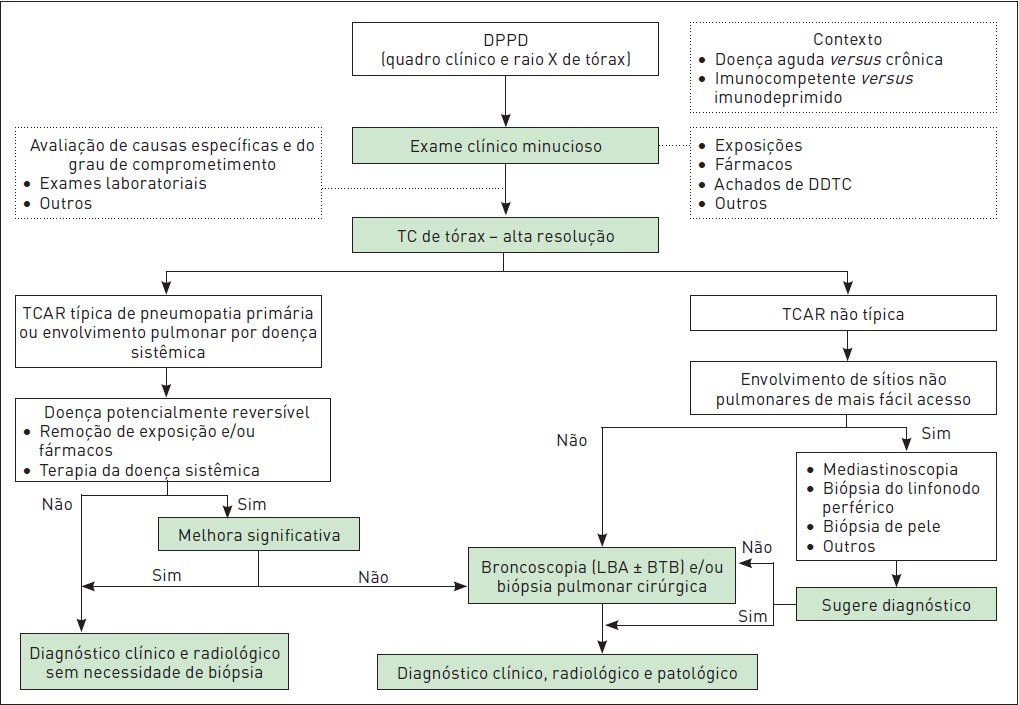
O Quadro 105.3 e a Figura 105.4 destacam os aspectos fundamentais na abordagem das DPPDs.
O tratamento das DPPDs é bastante variável, conforme a etiologia específica, seja relacionada à doença pulmonar de etiologia específica (p. ex., antifúngicos para paracoccidioidomicose) ou ao tratamento da doença sistêmica com envolvimento pulmonar (p. ex., quimioterapia para linfangite carcinomatosa por câncer de mama, antimicobacterianos na tuberculose miliar, imunomoduladores no envolvimento pulmonar por DDTC). No entanto, algumas recomendações gerais podem ser feitas.26
Abolir, ou pelo menos reduzir, as exposições é o tratamento principal para pacientes com doenças relacionadas às exposições. Também é importante sua realização em casos de outras doenças, a fim de evitar lesão adicional à doença de base. A maioria dos pacientes com PH apresenta resolução completa (se a doença é aguda ou subaguda) ou, pelo menos, estabilização após cessada a exposição, sendo que apenas uma parcela necessita de tratamento farmacológico adicional. Nos casos de pneumoconioses, também a interrupção da exposição evita que a doença progrida de forma mais rápida, embora o metal já inalado não possa ser depurado e continue a causar lesão pulmonar. A cessação do tabagismo é essencial para todos os pacientes, e para aqueles com doenças relacionadas ao tabaco (p. ex., pneumonia intersticial descamativa) pode conduzir à melhora parcial das lesões em alguns casos. Nas pneumopatias induzidas por fármacos, a suspensão do medicamento em geral gera resolução do quadro, a menos que ele já seja crônico (p. ex., fibrose por amiodarona), caso em que, então, a interrupção evita lesão adicional. Em raros casos, não é possível suspender o fármaco (doença grave e sem alternativa terapêutica), devendo o paciente utilizar a menor dose possível e tratar a pneumopatia concomitantemente (em geral com corticoide sistêmico).
Figura 105.4
Abordagem das doenças pulmonares parenquimatosas difusas.
LBA, lavado broncoalveolar; BTB, biópsia transbrônquica.
Várias medidas são importantes. É fundamental evitar lesões pulmonares adicionais, sendo, portanto, fundamental a cessação absoluta do tabagismo. Além disso, a realização de vacinas para influenza e pneumococo pode reduzir a incidência de doenças infecciosas associadas. O suporte nutricional visa a evitar desnutrição e sobrepeso. A incidência de depressão e ansiedade é elevada, sendo relevante o acompanhamento psicológico e/ou psiquiátrico. O tratamento das comorbidades, que são frequentes (p. ex., diabetes, hipertensão arterial, cardiopatia isquêmica), pode melhorar a qualidade de vida dos pacientes. A osteoporose é comum em pacientes que utilizam cronicamente corticoide, sendo importante o uso preventivo de cálcio, vitamina D e, de forma eventual, bifosfonados ou outros inibidores da reabsorção óssea. Também se deve efetuar o manejo sintomático do cor pulmonale secundário à pneumopatia com diuréticos, eventualmente digitálicos e raramente sangria terapêutica. A terapia com fármacos específicos para hipertensãopulmonar(inibidores da fosfodiesterase, antagonistas da endotelina e análogos das prostaglandinas), em pacientes com DPPD, ainda não está comprovada. Já que muitos pacientes com DPPD utilizam imunossupressores, devem-se administrar periodicamente anti-helmínticos para evitar uma infestação maciça (p. ex., estrongiloidíase sistêmica), realizar quimioprofilaxia para casos de tuberculose, quando indicada, e profilaxia com cotrimoxazol (a menos que haja alergia à sulfa) para pacientes com pneumocistose que utilizem esquemas mais imunossupressores (p. ex., corticoide associado à ciclofosfamida).
Embora não haja estudos específicos realizados em pacientes com DPPD, seguem-se as mesmas indicações de oxigenoterapia dos pacientes com DPOC (PaO2 = 55 mmHg ou SatO2 =88%, ou presença de cor pulmonalecom PaO2 56 a 59 mmHg ou SatO2 de 89%).
Programas de reabilitação pulmonar são menos frequentes para pacientes com DPPD do que para pacientes com DPOC e geralmente são subutilizados. A falta de condicionamento físico é frequente, como em outras pneumopatias crônicas, e está associado à limitação das atividades da vida diária e da qualidade de vida. Deve-se adaptar o nível de treinamento às condições dos pacientes. Recomenda-se o uso de oxigenoterapia adicional para os que dessaturam durante o exercício.
Além dos exercícios físicos, o programa de reabilitação engloba aspectos nutricionais, psicológicos e educativos. A integração entre os participantes auxilia os pacientes no entendimento e na aceitação de sua doença. Os programas em geral duram de 8 a 12 semanas, seguidos de um período de manutenção a longo prazo.
Corticoides. Os corticoides sistêmicos são os fármacos mais utilizados para o tratamento de muitas DPPDs, tendo em vista o componente inflamatório presumido. No manejo de pacientes ambulatoriais, utiliza-se prednisona, 0,5 a 1 mg/kg/dia, (ou equivalente) em dose única diária.Deve-se reduzir essa dose progressivamente até que se consiga estabelecer a mínima dose possível (p. ex., 10 mg ao dia) ou idealmente até a sua suspensão. Em casos de doenças como a sarcoidose, o corticoide pode ser utilizado em dias alternados. Os efeitos adversos são bem conhecidos: hiperglicemia, hipertensão, osteoporose, catarata, fragilidade cutânea, obesidade, infecções, entre outros. As medidas específicas de monitoração e terapia para pacientes com essas condições devem ser realizadas paralelamente. Nos pacientes com agudização de DPPD, às vezes se utiliza pulsoterapia com corticoide (500 a 1.000 mg ao dia, por 3 a 5 dias). No Quadro 105.4, estão discrimidas as doenças que apresentam resposta favorável e desfavorável à corticoterapia.2,25
Os corticoides inalatórios são menos administrados, sendo empregados em geral quando há hiper-responsividade brônquica associada às DPPDs (p. ex., na sarcoidose).
Imunossupressores. Os imunossupressores (não corticoides) mais utilizados nas DPPDs são a azatioprina, a ciclofosfamida e o metotrexato, cuja opção depende do contexto clínico.27,28 Na FPI, a rara e discutível resposta ao tratamento imunossupressor pode demorar três a seis meses, embora seja possível que os sintomas melhorem antes. Havendo estabilidade ou melhora gradual das alterações funcionais, o tratamento deve ser estendido por, no mínimo, 18 meses.9
Administra-se a azatioprina na dose diária de 1 a 2 mg/kg de peso, não excedendo 150 mg ao dia. Inicia-se com 25 a 50 mg ao dia, aumentando progressivamente 25 mg a cada uma a duas semanas, monitorando o hemograma e as provas de função hepática. Os principais efeitos adversos são gastrintestinais (náuseas, vômitos, dispepsia), hepatotoxicidade e supressão medular (leucopenia e plaquetopenia). O potencial de indução de neoplasia é controverso.
Utiliza-se ciclofosfamida na dose diária de 1 a 2 mg/kg de peso, não ultrapassando 150 mg ao dia. Deve-se iniciar com 25 a 50 mg ao dia, ingeridos pela manhã (orientando o paciente a urinar em até quatro horas após a dose), e aumentar 25 a 50 mg ao dia a cada duas semanas. Um esquema alternativo é a pulsoterapia mensal, na dose de 500 a 750 mg/m2, intravenosa. Os principais efeitos adversos são hematológicos, gastrintestinais, alopecia, hepatotoxicidade, pneumonite, cistite hemorrágica, exacerbação de doenças virais e neoplasias secundárias. Deve-se estimular os pacientes a manter uma ingesta hídrica adequada e a vigiar a ocorrência de hematúria. No uso intravenoso, recomendam-se geralmente hidratação vigorosa e pré-medicação com mesna, a fim de evitar cistite hemorrágica. Devem-se realizar hemograma e exame de urina antes do tratamento, quando são realizadas modificações de dose, e, depois, periodicamente. A dose cumulativa de ciclofosfamida não deve exceder 50 a 70 g, uma vez que há uma forte associação com o desenvolvimento de neoplasias, devendo o esquema imunossupressor ser feito com outra medicação.
O metotrexato é um imunossupressor utilizado com frequência em casos de artrite reumatoide. Também é administrado para pacientes com sarcoidose quando há intolerância ou falha da corticoterapia. Prescreve-se uma dose semanal inicial de 7,5 mg, aumentando-se 2,5 mg por semana até que se atinja uma dose máxima de 25 mg por semana. Recomenda-se a suplementação de ácido fólico de 5 mg, uma a duas vezes por semana. Os efeitos adversos mais comuns são alterações hematológicas, gastrinterstinais, hepáticas, pneumonite e úlceras orais. Há associação com o desenvolvimento de fibrose ou cirrose hepática, sendo que, em algumas diretrizes, embora haja controvérsia, recomenda-se a realização de biópsia hepática de vigilância quando a dose cumulativa atinge 1,5 a 2,5 g ou a cada três anos.
Outros imunossupressores (p. ex., micofenolato, rituximabe, ciclosporina, sirolimus) podem ser utilizados em situações particulares, embora as evidências estejam geralmente embasadas em casos anedóticos.
Outros fármacos. A N-acetilcisteína (NAC) é um antioxidante que aumenta os níveis de glutationa, reduzindo a liberação de radicais livres de oxigênio, os quais estão envolvidos, pelo menos parcialmente, na patogênese da FPI. Em um ensaio clínico, observou-se que a NAC, na dose de 600 mg, três vezes ao dia, associada a um esquema imunossupressor (prednisona e azatioprina), promoveu um declínio mais lento da CVF e da DCO em relação ao grupo que utilizou somente o esquema imunossupressor.29
Medicamentos antifibróticos têm sido testados para o tratamento das doenças pulmonares intersticiais (DPIs) fibrosantes, principalmente a FPI. Entre eles, estão a colchicina, a penicilamina e o interferon gama-1b, porém os resultados não evidenciaram eficácia terapêutica. O fármaco mais promissor é a perfenidona, que apresenta ações antifibróticas, antioxidantes e anti-inflamatórias.
Outros fármacos com mecanismos de ações diversos já foram testados no tratamento da FPI e não mostraram benefício, como a bosentana, o etanercepte e o imatinibe.
Embora seja questionado, o tratamento com medicamentos empíricos do refluxo gastresofágico tem sido recomendado para pacientes com FPI, tendo em vista a elevada prevalência e a possibilidade de eficácia terapêutica.30
Deve ser considerado para pacientes com menos de 65 anos, declínio funcional progressivo e piora da dispneia (apesar de tratamento otimizado), hospitalizações repetidas por doença respiratória ou avaliação inicial que indique doença avançada (CVF menor do que 60% do previsto, DCO menor do que 40% do previsto, queda da SpO2 para menos de 90% com exercício moderado e extensa fibrose na TCAR). É essencial a inexistência de disfunção significativa em outros órgãos e um perfil psicossocial adequado.11,30
A exacerbação de uma DPPD é um contexto potencial de alta mortalidade. Pode ocorrer tanto em casos de FPI quanto de outras doenças pulmonares intersticiais idiopáticas ou associadas à DDTC. O manejo em geral envolve a exclusão de complicações, como pneumonia, tromboembolismo pulmonar e pneumotórax, ou descompensação de comorbidades, como insuficiência cardíaca ou coronariopatias. Geralmente o tratamento específico da exacerbação implica aumento da imunossupressão, principalmente da corticoterapia (seja por aumento da dose diária para 1 a 2 mg/kg ao dia ou por pulsoterapia com 0,5 a 1 g de metilprednisolona ao dia, durante três a cinco dias). Em alguns casos, como nos de síndromes pulmão-rim (principalmente de granulomatose de Wegener e síndrome de Goodpasture), pode haver benefício da plasmaférese. No contexto da FPI, o prognóstico é particularmente grave, atingindo mortalidade intra-hospitalar de até 65% e de mais de 90% em seis meses.22,31 Nos indivíduos que necessitam de suporte ventilatório invasivo, a taxa de mortalidade aproxima-se de 100%, sendo recomendado discutir com o paciente e os familiares o risco-benefício dessa terapia, considerando o prognóstico a curto e a médio prazos.
Devem-se avaliar periodicamente os pacientes com DPPD, em geral, a cada três a seis meses, dependendo da doença e de sua gravidade. Nas consultas, questiona-se sobre sintomas respiratórios, limitações na vida diária (escala de dispneia e classe funcional), hospitalizações, adesão à terapia, efeitos adversos dos medicamentos, aspectos nutricionais e psicológicos, o status tabágico (mesmo se já parou de fumar). Realizam-se os exames de controle também conforme a doença de base. Efetuam-se os exames laboratoriais (hemograma, função renal, hepática e eletrólitos) para monitoração de alguns medicamentos ou de doenças em que há alteração significativa (p. ex., cálcio sérico e urinário na sarcoidose). O teste tuberculínico deve ser executado em pacientes não reatores, antes do início da imunossupressão, ou naqueles com doenças que predispõem à tuberculose (p. ex., silicose), mesmo que sem tratamento, a fim de avaliar a necessidade de tratamento da tuberculose latente (uso de isoniazida durante seis meses). O exame de imagem, como raio X de tórax, é realizado periodicamente, e a TCAR, quando há dúvida na evolução que justifique modificação terapêutica (considerar que a exposição à radiação é alta nas TCs). Os testes de função pulmonar também são repetidos geralmente a cada três a seis meses, como espirometria antes e após o uso de broncodilatador, capacidade de difusão pulmonar e teste de caminhada de seis minutos (gasometria arterial se a SpO2 em ar ambiente for menor do que 93%). Realizam-se os exames cardiológicos (ECG, ecocardiograma e, em alguns casos, Holter e cateterismo cardíaco) quando haver suspeita de envolvimento cardíaco ou, pelo menos, anualmente nos indivíduos com DPPD mais avançada. Recomenda-se uma revisão oftalmológica periódica para pacientes com sarcoidose, para os que utilizam medicações tóxicas (p. ex., cloroquina) ou que têm comorbidades específicas (p. ex., diabetes).14
O prognóstico de pacientes com DPPDs varia principalmente de acordo com a etiologia específica. No protótipo das DPPDs, que é a FPI, o prognóstico é extremamente grave, alterando a sobrevida média entre dois e três anos, e somente 20 a 30% dos pacientes continuam vivos após cinco anos do diagnóstico. Em geral, há fatores comuns que apresentam implicação prognóstica, como o grau de comprometimento funcional (sobretudo de capacidade vital forçada, capacidade de difusão pulmonar, hipoxemia em repouso e desempenho nos testes de exercício, tais como distância percorrida no teste de caminhada de seis minutos e presença de dessaturação), a intensidade das alterações radiológicas (principalmente as que indicam doença avançada, como a existência de faveolamento), os sinais clínicos ou hemodinâmicos de cor pulmonale e envolvimento multissistêmico pela doença de base (sistema cardiovascular e neurológico). Para algumas doenças, como a FPI, existem escores prognósticos que englobam variáveis independentes relacionadas à sobrevida.32 A evolução das DPPDs não é invariavelmente de deterioração progressiva, podendo observar-se que a ocorrência de exacerbações leva a um rápido declínio na curva de sobrevida.
As bronquiolites são doenças que envolvem a via aérea periférica (bronquíolos), podendo ou não também comprometer o espaço alveolar. As etiologias são diversas: infecciosas (micoplasma, vírus), fármacos, DDTC, rejeição a transplante pulmonar e idiopáticas (POC, bronquiolite folicular, bronquiolite constritiva). Caracteristicamente apresentam um padrão obstrutivo, mas, em alguns casos, pode haver restrição por colabamento precoce da via aérea. Evidenciam-se radiologicamente nódulos centrolobulares e alçaponamento aéreo nos cortes em expiração e padrão de árvore em brotamento quando há lesão alvoelar associada. Direciona-se o diagnóstico para a etiologia específica, geralmente envolvendo realização de broncoscopia para LBA e eventualmente biópsia transbrônquica (BTB). O tratamento também é relacionado à doença de base ou à corticoterapia/imunossupressores nos casos idiopáticos.
O CBA, ou carcinoma de células alveolares, é um subtipo de adenocarcinoma e corresponde a menos de 10% das neoplasias pulmonares primárias.25 O tipo A é uma forma de doença difusa e deve ser diferenciada de metástases de adenocarcinoma de mama, ovário, pâncreas e estômago. O tipo B secreta mais mucina do que o tipo A e manifesta-se de forma mais localizada (nódulo ou lesão focal). Os sintomas são dispneia, tosse seca e perda de peso. A broncorreia ocorre em alguns casos. O padrão em “pavimentação maluca”, isto é, infiltrado em vidro fosco associado a espessamento dos septos interlobulares, é um achado sugestivo (Fig. 105.5). Deve-se considerar esse diagnóstico no contexto de uma pneumonia não resolvida. O LBA apresenta resultado positivo em muitos casos, já que as células ocupam os espaços alveolares. A resposta à quimio e à radioterapia era insatisfatória no passado, mas os novos esquemas farmacológicos evidenciam mais benefício.
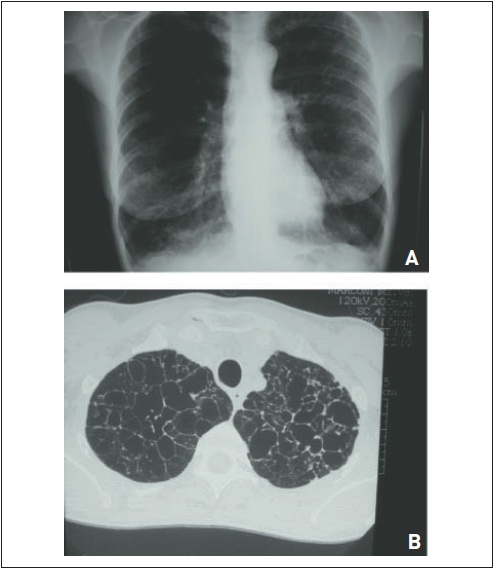
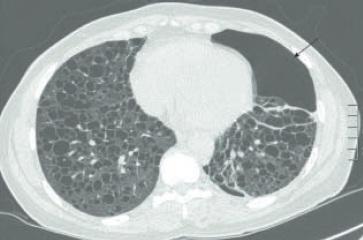
As doenças típicas são a linfangioleiomiomatose e a histiocitose pulmonar de células de Langerhans (granuloma eosinofílico). Essas condições ocorrem de forma rara e afetam, respectivamente, mulheres em idade fértil e homens jovens. A maioria dos pacientes com histiocitose é tabagista. As manifestações clínicas são variáveis, mas a ocorrência de pneumotórax deve lembrar essas doenças. Em casos de linfangioleiomiomatose, os cistos são regulares e, nos de histiocitose, são irregulares (bizarros) e com nódulos associados (Fig. 105.6 e 105.7). O envolvimento renal com forma de angiomiolipomas sugere, no contexto de doença cística pulmonar, o diagnóstico de linfangioleiomiomatose. O tratamento da histiocitose implica cessação do tabagismo e realização de quimioterapia nos casos que progridem. A terapia da linfangioleiomiomatose envolve o uso de sirolimus, na dose de 2 mg ao dia, com manutenção do nível sérico entre 5 e 15 ng/mL.22 No passado, utilizava-se progesterona ou tamaxifeno, embora com base em séries de casos.
Figura 105.5
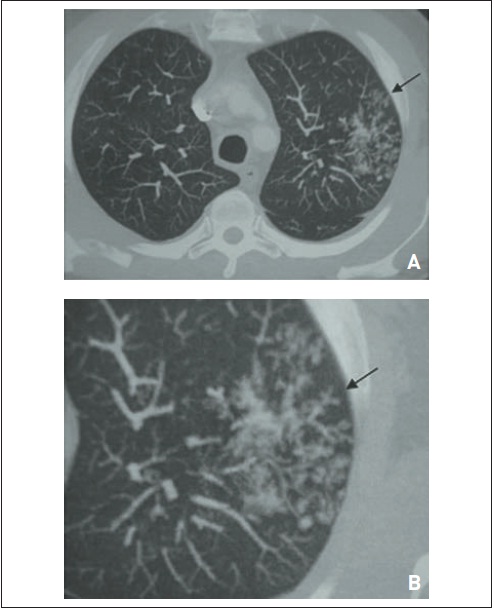
A e B – Adenocarcinoma bronquioloalveolar mucinoso. Há infiltrado pulmonar difuso em vidro fosco com espessamento dos septos interlobulares, denominado “pavimentação maluca” (seta).
O exemplo mais comum dessas doenças são as que causam hemorragia alveolar, tais como síndrome de Goodpasture, hemossiderose pulmonar, algumas vasculites pulmonares ou pneumopatias por drogas (Fig. 105.8). Nesses casos, os sintomas são dispneia e hemoptise, embora em até um terço dos pacientes possa não ser evidente exteriorização do sangramento, havendo somente o achado de anemia ferropriva. Radiologicamente, há um infiltrado alveolar, caracterizado por opacidades imprecisas e coalescentes (Fig. 105.9). Em casos de síndrome de Goodpasture, frequentemente ocorre envolvimento renal (faz parte do espectro das síndromes pulmão-rim). O anticorpo antimembrana basal subsidia o diagnóstico, que é confirmado por achado típico de depósito de imunocomplexos na membrana basal glomerular ou alveolar. O tratamento principal é a plasmaférese e a corticoterapia. Em alguns casos, pode haver necessidade de uso de imunossupressores, em geral ciclofosfamida ou azatioprina. Outros exemplos de doenças em que ocorre comprometimento alveolar são proteinose alveolar, CBA, pneumocistose, pneumonias infecciosas e reação a fármacos.
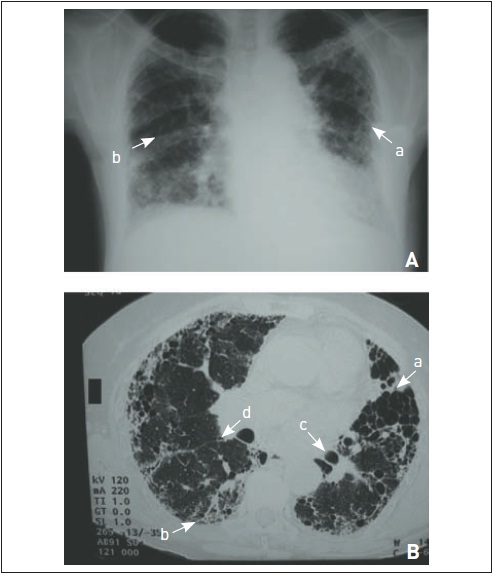
Figura 105.6
Histiocitose X pulmonar (granuloma eosinofílico). Múltiplos cistos de formatos irregulares são característicos dessa doença.
Figura 105.7
Linfangioleiomiomatose. Há cistos de paredes regulares em vários tamanhos e pneumotórax à esquerda (seta).
Em geral o envolvimento pulmonar nas DDTCs ocorre concomitantemente ou durante a evolução da doença, embora, em cerca de 10% dos casos, possa não haver outras manifestações extrapulmonares (colagenoses ocultas).33 Os sintomas mais comuns são dispneia e tosse seca, associados às manifestações reumáticas. As apresentações radiológicas variam conforme a doença de base, podendo mais frequentemente apresentar padrão consolidativo (em casos de LES com pneumonite ou hemorragia alveolar) ou pneumonite intersticial em geral de padrão não específico (em casos de esclerose sistêmica, artrite reumatoide e polimiosite) (Fig. 105.10). Se o diagnóstico da DDTC está estabelecido e o padrão radiológico é característico, não é necessária a realização de biópsia pulmonar. Confirma-se o diagnóstico por meio de uma combinação de características clínicas e de autoanticorpos específicos para a doença em questão. O tratamento envolve corticoterapia, uso de imunossupressores e agentes biológicos.
Figura 105.8
Exemplos de doença pulmonar parenquimatosa difusa de apresentação aguda. (A) Leptospirose com envolvimento pulmonar; (B) Pneumocistose.
Essas doenças compõem um grupo em que há uma infiltração por eosinófilos. As manifestações principais são envolvimento pulmonar por parasitas (síndrome de Loeffler), reações a drogas, síndrome de Churg-Strauss, aspergilose broncopulmonar alérgica e pneumonias eosinofílicas idiopáticas, agudas ou crônicas, e síndrome hipereosinofílica com envolvimento pulmonar. Não ocorre eosinofilia periférica em todos os casos. Os sintomas são tosse, sibilância e dispneia, mas variam entre as doenças. O padrão radiológico é diverso, podendo ser focal ou difuso, alveolar, intersticial ou misto. O padrão de consolidações periféricas (edema pulmonar negativo) sugere pneumonia eosinofílica crônica. Para o diagnóstico, realizam-se LBA e biópsia pulmonar (broncoscópica ou cirúrgica), sendo obviamente de exclusão nas formas idiopáticas. Direciona-se o tratamento para a doença de base, e efetua-se corticoterapia em pacientes com formas de causa desconhecida.
Figura 105.9
Síndrome de Goodpasture com hemorragia alveolar. Observa-se infiltrado em padrão alveolar com opacidades imprecisas, coalescentes e que poupam as regiões subpleurais (setas).
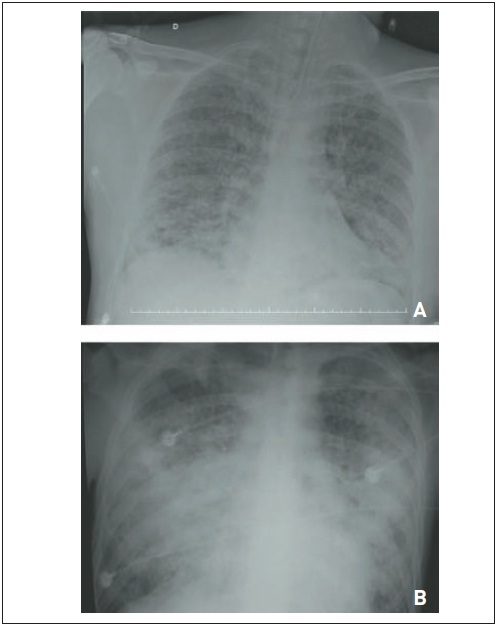
O edema pulmonar, em geral de apresentação aguda, pode evidenciar padrão hidrostático, nefrogênico ou cardiogênico (relacionado à insuficiência cardíaca diastólica ou sistólica ou a valvopatias, como estenose mitral) ou não hidrostático ou inflamatório, que, nos casos agudos, manifestam-se por lesão pulmonar aguda (e, nos mais graves, síndrome do desconforto respiratório agudo, com relação PaO2/FiO2 menor do que 200). No edema hidrostático, que ocorre devido ao aumento do volume intravascular pela diferença de pressão, há cardiomegalia, derrame pleural bilateral e infiltrado intersticial que pode progredir para comprometimento alveolar (edema agudo de pulmão) (Fig. 105.11). Na radiografia de tórax, verifica-se comprometimento alveolar difuso nos quadros não hidrostáticos (que ocorrem por aumento da permeabilidade da membrana alveolocapilar), embora os critérios radiológicos não apresentem especificidade satisfatória. O diagnóstico definitivo para diferenciar causas cardíacas de não cardíacas é estabelecido por meio da cateterização da artéria pulmonar, que, nos casos não cardiogênicos, evidencia baixas pressões de enchimento (pressão de oclusão menor do que 18 mmHg). Para estabelecer o diagnóstico etiológico, executam-se exames como TC de tórax, ecocardiograma, dosagem de peptídeo natriurético, entre outros. O tratamento envolve o manejo da doença de base.
Figura 105.10
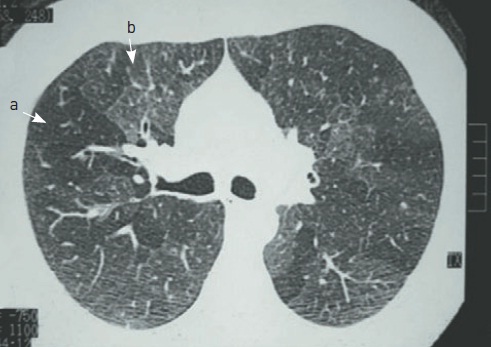
Pneumonite intersticial não específica associada à esclerose sistêmica. Há infiltrado tipo vidro fosco homogêneo em bases, espessamento de septos interlobulares e das cissuras, algumas bronquiectasias de tração, mas não se observa faveolamento.
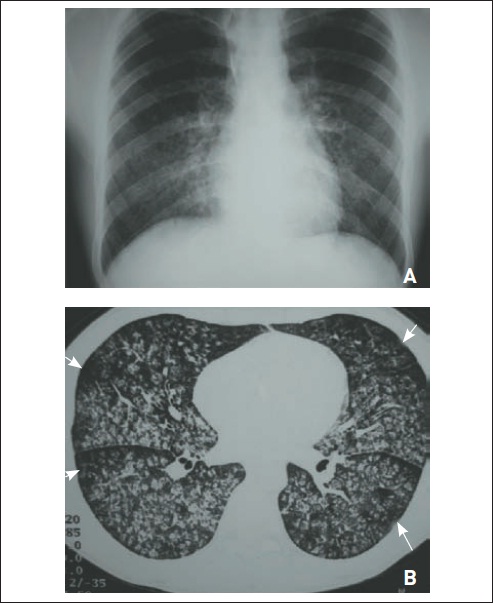
A FPI corresponde a cerca de 50 a 60% de todas as pneumonites intersticiais idiopáticas.9 Ocorre mais comumente em homens após os 50 anos de idade, muitos dos quais são tabagistas ou já foram. Há hipocratismo digital em 25 a 50% dos casos. O quadro típico é de dispneia progressiva, tosse seca, crepitantes em velcro nos campos pulmonares inferiores, espirometria com padrão restritivo, capacidade de difusão pulmonar reduzida e, nos exames de imagem, a verificação de infiltrado reticular que predomina nas bases e nas regiões subpleurais, associado a áreas de faveolamento. Recentemente, a American Thoracic Society revisou os critérios diagnósticos, sugerindo critérios tomográficos e histológicos e suas combinações, que devem ser discutidos em reunião multidisciplicar com o clínico, o radiologista e o patologista.30 Em pacientes com achados típicos na TCAR, excluídas outras causas (em geral, exposições, DDTC e fármacos), é possível confirmar o diagnóstico (Fig. 105.12). Já nos indivíduos em que a TCAR não é típica, faz-se necessário realizar biópsia pulmonar cirúrgica. Não se recomenda a execução de broncoscopia quando a suspeita é de FPI, sendo indicada quando a principal suspeita é uma doença cujo rendimento do exame é maior (p. ex., sarcoidose, linfangite carcinomatosa). O achado histopatológico característico é o padrão de pneumonite intersticial usual, em que há focos fibroblásticos, deposição de colágeno, faveolamento e pouco componente inflamatório e, nos casos não avançados, preservação de algumas áreas.34 Pode-se observar esse padrão em algumas DDTCs e pneumopatias por fármacos. O prognóstico é reservado, e a sobrevida média é de dois a três anos.
As terapias atuais auxiliam pouco. Pode haver discreto e discutível benefício com o uso de N-acetilcisteína,anticoagulação plena, combinação de corticoide, imunossupressor e N-acetilcisteína e tratamento de refluxo gastresofágico (mesmo que assintomático). O uso de corticoide sistêmico isoladamente é contraindicado. Deve-se discutir com o paciente o risco-benefício do tratamento. Alguns pacientes, geralmente os tabagistas, podem apresentar enfisema e fibrose pulmonar concomitantes, sendo caracterizados por dispneia intensa, espirometria com poucas alterações (desproporcionais aos sintomas), redução grave da capacidade de difusão, dessaturação ao exercício e sinais de hipertensão pulmonar significativa. As exacerbações da FPI apresentam mortalidade elevadíssima e podem ser tratadas com pulsoterapia com corticoide, sendo necessário descartar causas infecciosas e tromboembólicas da agudização.
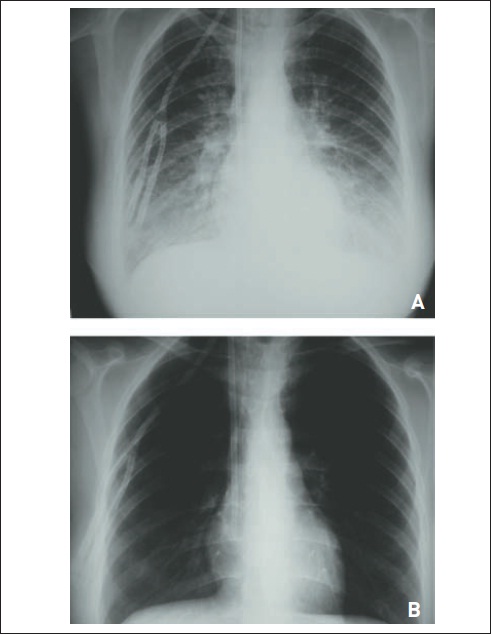
Figura 105.11
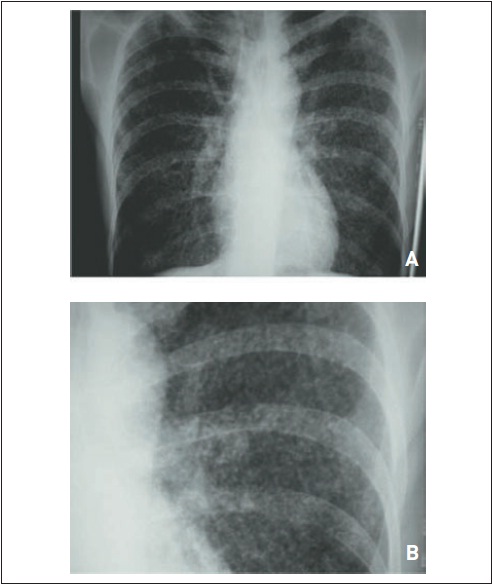
Edema pulmonar hidrostático. (A) Paciente com insuficiência renal crônica que apresenta infiltrado pulmonar difuso, principalmente em lobos inferiores, caracterizando edema pulmonar. (B) Essas alterações foram tratadas após uma sessão de hemodiálise (observar o cateter de Schilley).
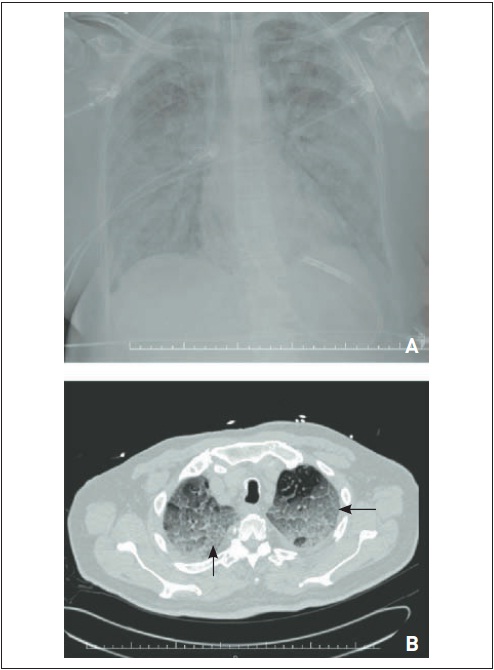
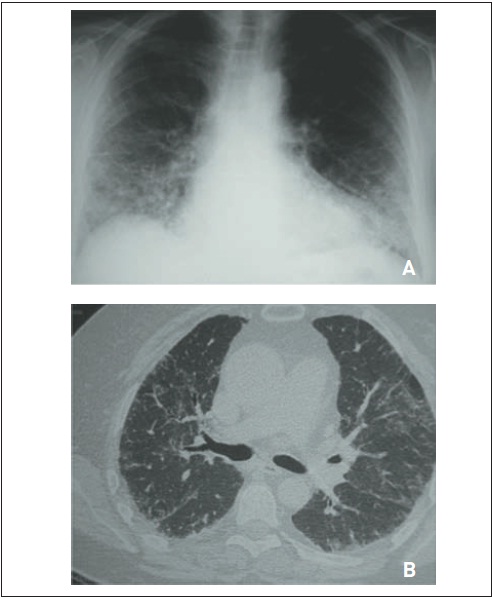
Figura 105.12
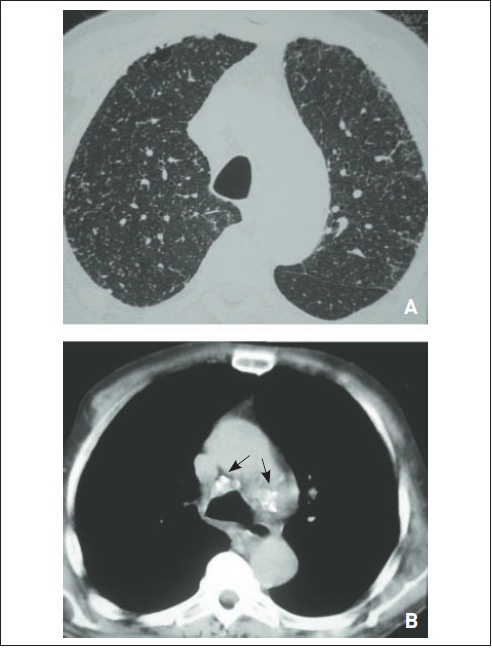
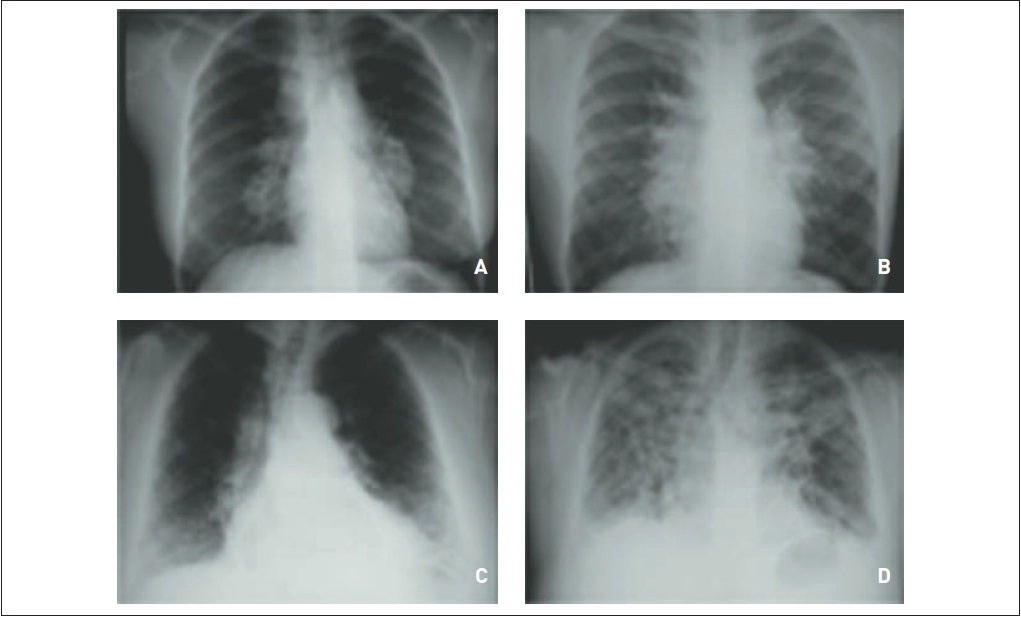
Fibrose pulmonar idiopática. RaioX (A) e tomografia computadorizada de tórax (B) evidenciam (a) áreas de faveolamento subpleural predominando em lobos pulmonares inferiores, (b) infiltrado intersticial de padrão septal, (c) bronquiectasias de tração e (d) espessamento de cissuras.
c
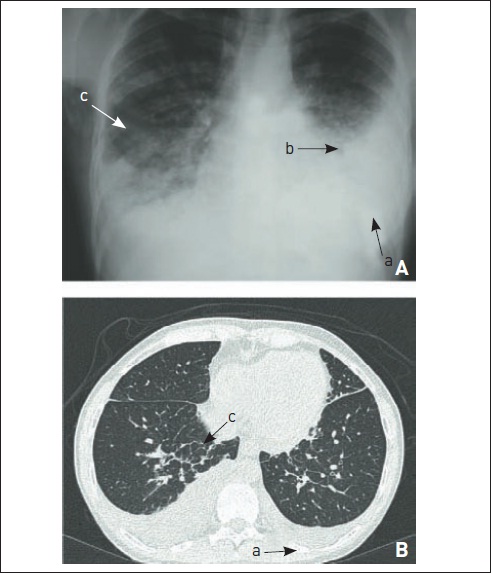
Figura 105.13
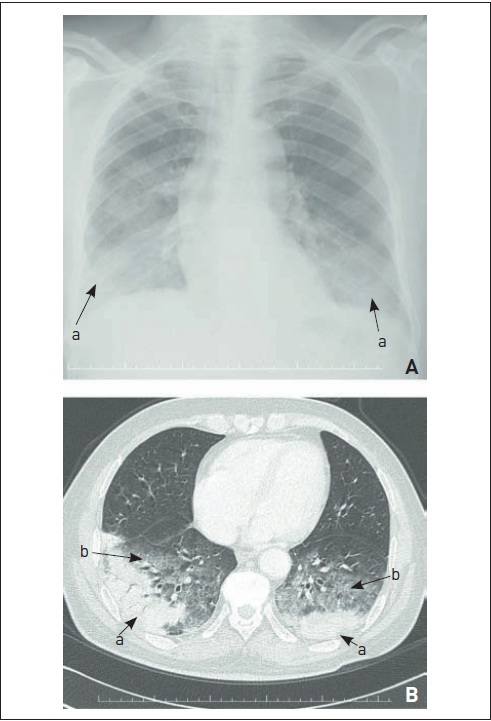
Linfangite carcinomatosa.Observam-se (a) derrame pleural, (b) adenomegalias mediastinais e (c) infiltrado intersticial septal com espessamento dos septos interlobulares.
A linfangite carcinomatosa é uma forma-padrão de metástase pulmonar que apresenta envolvimento intersticial nos exames de imagem do tórax. As neoplasias mais frequentemente associadas são câncer de mama, pulmão, estômago, pâncreas, próstata, ovário e rim. Raramente, pode haver sarcomas ou carcinomas escamosos de colo uterino ou orofaringe nesse padrão. O sintoma principal é a dispneia progressiva, embora, em alguns casos, possa ocorrer também insuficiência respiratória aguda em neoplasias agressivas. A ocorrência de tosse irritativa também é comum. Tipicamente, há progressão para cor pulmonale e óbito em três a seis meses. Nos exames de imagem, observam-se infiltrado intersticial reticular nas bases e linhas B de Kerley, os quais geralmente evoluem para doença extensa. São comuns derrame pleural e adenomegalias mediastinais (Fig. 105.13). Na maioria dos casos, a neoplasia já é conhecida. É importante realizar o diagnóstico diferencial com toxicidade pelos quimioterápicos ou radioterapia (pneumonite actínica), bem como complicação infecciosa e insuficiência cardíaca decorrente de cardiotoxicidade. Estabelece-se o diagnóstico geralmente por meio de LBA e biópsia pulmonar broncoscópica. Embora o diagnóstico seja grave na maioria dos casos, nas neoplasias de próstata e mama pode haver boa resposta e benefício em sobrevida.
As micoses pulmonares mais frequentes são a paracoccidioidomicose, a histoplasmose, a aspergilose e a criptococose. Os sintomas e as alterações radiológicas são variadas. A paracoccidioidomicose, que ocorre frequentemente em homens agricultores, está associada a lesões orais e infiltrados reticuloalveolares que predominam nos terços médios e nos superiores. O envolvimento pulmonar, em casos de histoplasmose, pode ser focal (lesão pulmonar primária) ou difuso (doença sistêmica). A aspergilose é comum em pacientes imunodeprimidos (aspergilose invasiva) ou em pneumopatas (forma necrosante crônica). No contexto clínico apropriado, a verificação de sinal do halo na tomografia (consolidação circundada por vidro fosco) sugere acentuadamente aspergilose angioinvasiva. Realiza-se o diagnóstico por exame de escarro, LBA ou biópsia pulmonar ou de lesões extrapulmonares. O antígeno sérico galactomanana pode indicar o diagnóstico de aspergilose invasiva. O tratamento implica o uso de antifúngicos imidazólicos (itraconazol, fluconazol, voriconazol), anfotericina e estreptogranina.
As pneumoconioses são doenças geralmente ocupacionais relacionadas à inalação de poeira ou partículas de substâncias inorgânicas. As substâncias mais comumente envolvidas são sílica, asbesto, carvão, berílio, silicatos e poeira mista. Há profissões particularmente associadas às pneumoconioses (Tab. 105.4). Dependendo da frequência de exposição e da reação dos indivíduos, pode ocorrer uma acentuada latência (mais de 10 anos) entre a exposição e o aparecimento de sintomas. Além de utilizar sempre equipamentos de proteção individual, os indivíduos expostos a essas substâncias devem realizar radiografias de tórax e espirometrias anuais e ser afastados se houver indício de pneumopatia. O quadro em geral é crônico, e o sintoma predominante é a dispneia, mas a exposição muito frequente pode levar a um padrão de síndrome da angústia respiratória aguda. Na maioria dos casos, estabelece-se o diagnóstico de pneumoconiose por meio da história de exposição associada a achados radiológicos ou tomográficos compatíveis, não havendo necessidade de realização de biópsia pulmonar.
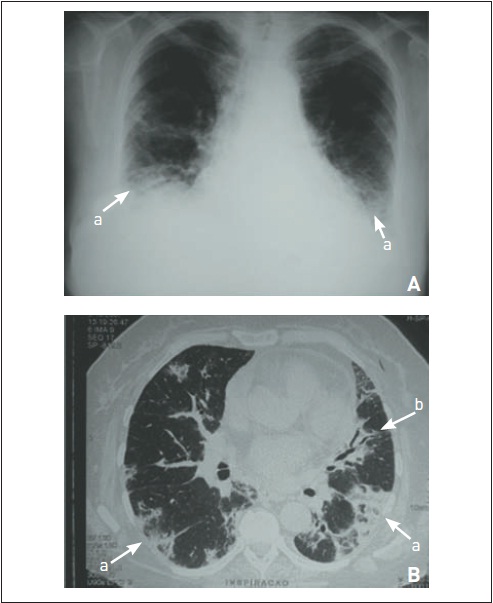
Em pacientes com silicose, há nódulos parenquimatosos principalmente nos campos pulmonares superiores e que podem aglomerar-se com a evolução da doença (fibrose maciça), além de adenomegalias mediastinais calcificadas (em casca de ovo) (Fig. 105.14). Em casos de asbestose, ocorre infiltrado intersticial semelhante ao da FPI, além de envolvimento pleural (espessamento, placas calcificadas). A Organização Internacional do Trabalho recomenda uma classificação radiológica específica para as pneumoconioses, com base no tipo, no tamanho e no padrão de opacidades pulmonares. A pesquisa de cristais do LBA positivo indica somente exposição, e os achados anatomopatológicos, geralmente granulomas de corpo estranho, não são essenciais para o diagnóstico, sendo a biópsia realizada somente em casos atípicos. A exceção à regra é o diagnóstico de beriliose, que pode ser de realização muito difícil, principalmente quando a exposição ocupacional não é evidente, uma vez que os achados histopatológicos podem ser idênticos aos da sarcoidose.35 O método recomendado é o teste de proliferação de linfócitos periféricos estimulados pelo berílio, mas está disponível em poucos centros. O tratamento é o afastamento da exposição e a prevenção de lesões adicionais (p. ex., cessação do tabagismo e realização de quimoprofilaxia para tuberculose).
Figura 105.14
Silicose. Há múltiplos micronódulos difusos, às vezes coalescentes.Também se visualizam adenomegalias mediastinais calcificadas.
A PH, ou alveolite alérgica extrínseca, é uma doença granulomatosa causada pela inalação de substâncias orgânicas (ou inorgânicas de baixo peso molecular) que se ligam a haptenos. Os agentes mais comuns são o mofo domiciliar e as penas de pássaros. O quadro clínico pode apresentar-se de forma aguda, subaguda ou crônica, conforme o grau de exposição e a reação imunológica do indivíduo. Da mesma forma, a apresentação radiológica é relacionada ao tempo de instalação. Estabelece-se o diagnóstico por meio de biópsia, a qual evidencia granuloma de corpo estranho, embora achados típicos observados nos exames de imagem e a melhora após o afastamento da exposição sejam suficientes para um diagnóstico presuntivo. O tratamento, na maioria dos casos, é a remoção dos agentes inalados. A busca pela exposição dever ser incessante, uma vez que a doença pode evoluir para fibrose avançada. Em alguns casos, há necessidade de corticoterapia.
As PIIs são um subgrupo de DPIs agudas e crônicas, de etiologia desconhecida, caracterizadas pela existência de variados graus de inflamação alveolar-intersticial e fibrose. Esses padrões histológicos podem ser decorrentes de DDTC, fármacos, exposições e algumas infecções, sendo necessária a sua exclusão para confirmar realmente a forma idiopática. Nesse grupo, estão incluídas FPI, PINE, PID, doença pulmonar intersticial associada a bronquiolite respiratória (DPI-BR), pneumonite intersticial aguda (PIA), pneumonia em organização criptogênica (POC) e PIL. As manifestações da maioria das PIIs são dispneia progressiva ao esforço e tosse seca. Dor torácica e emagrecimento podem ocorrer. As alterações observadas no raio X e na TCAR de tórax são distintas entre as doenças. A PINE pode apresentar um padrão celular e um fibrótico, sendo o primeiro de melhor prognóstico e geralmente é o que responde ao uso de corticoide. A DPI-BR ocorre exclusivamente em fumantes e evidencia boa progressão após a cessação do uso do tabaco. Em sua forma mais extensa, ocorre em casos de PID, que caracteristicamente apresentam TCAR com perfusão em mosaico e biópsia com intenso acúmulo de macrófagos pigmentados intra-alveolares, hiperplasia de pneumócitos tipo II, espessamento dos septos e mínima fibrose (Fig. 105.15). A PIA, também denominada síndrome de Hamman-Rich,representa uma situação grave, que mimetiza SDRA e ocorre em geral após uma infecção respiratória viral. A PIL é quase exclusivamente associada à DDTC (em especial síndrome de Sjögren), apresentando quadro tomográfico típico de vidro fosco bilateral, nódulos centrolobulares e cistos. A POC, ante- riormente chamada de BOOP (bronquiolite obliterante com pneumonia em organização), é uma doença sem causa que se manifesta com um quadro similar ao de influenza, com tosse seca, seguido de dispneia (Fig. 105.16). Na TCAR, verificam-se típicas áreas de consolidação e vidro fosco subpleurais ou peribrônquicas. Os pacientes podem apresentar padrão funcional obstrutivo, embora o restritivo seja mais comum. Os achados observados na biópsia são de proliferação excessiva de tecido granular dentro da via aérea, dos alvéolos e ao seu redor. As recorrências são comuns, e a remissão espontânea também pode ocorrer. A resposta ao uso de corticoide em geral é satisfatória, devendo sua administração ser mantida durante, no mínimo, seis meses.
Figura 105.15
Pneumonite intersticial descamativa. Padrão de perfusão em mosaico, alternando (a) áreas hipodensas e (b) áreas de vidro fosco.
Figura 105.16
Pneumonia em organização criptogênica. Observam-se (a) consolidações subpleurais com bronco grama aéreo em regiões dorsais dos pulmões e (b) áreas adjacentes de vidro fosco.
Mais de 300 fármacos, biomoléculas e medicamentos homeopáticos podem causar pneumopatia aguda, subaguda ou crônica (Tab. 105.2).36 O mesmo fármaco pode causar diversos padrões radiológicos, dessa forma, o mesmo padrão pode ser causado por vários fármacos. O padrão histológico também é variável, incluindo pneumonia em organização, pneumonite granulomatosa, pneumonite intersticial não específica, entre outros. Os medicamentos que devem ser considerados tipicamente são a amiodarona, a nitrofurantoína e o metotrexato (Fig. 105.17). Estabelece-se o diagnóstico por meio da resolução das lesões após a suspensão do fármaco, embora a confirmação se dê pelo retorno das alterações quando há nova administração, o que raramente é feito. Em casos mais graves, pode ser necessária a realização de corticoterapia.
A sarcoidose é uma doença granulomatosa sistêmica de etiologia desconhecida. Afeta principalmente indivíduos jovens, com pico de incidência entre 20 e 30 anos.37,38 A sarcoidose pode comprometer qualquer órgão, mais comumente os pulmões, olhos (uveíte e ceratoconjuntivite seca) e pele (lúpus pérnio, nódulos subcutâneos, placas). O envolvimento neurológico (pares cranianos, principalmente o nervo facial, e encefalopatia, com predileção pelas estruturas da base do crânio) e cardíaco (bloqueios cardíacos, disfunção ventricular restritiva) denotam doença mais grave. Pode haver hipercalciúria e eventualmente hipercalcemia, as quais são causadas pela produção nos granulomas de derivados da vitamina D. A existência de sintomas e sinais agudos, como o eritema nodoso (síndrome de Loefgren), em geral se relaciona a um melhor prognóstico. Os sintomas mais comuns no envolvimento respiratório são dispneia e tosse. Hipocratismo digital e crepitantes em velcro são infrequentes. Na radiografia de tórax, há uma combinação de adenomegalias hilares bilaterais/simétricas e infiltrado em interstício axial peribrônquico. O estágio I caracteriza-se por envolvimento ganglionar mediastinal isolado, o II, por envolvimento mediastinal e parenquimatoso, o III, por acometimento parenquimatoso isolado, e o IV, por fibrose pulmonar (Fig. 105.18). O padrão radiológico indica probabilidade de remissão espontânea (p. ex., 60% no estágio I), não tendo significado evolutivo (i. é., o paciente não obrigatoriamente evolui do estágio I para o II e assim sucessivamente).9 Na, TCAR, os achados típicos são pequenos nódulos irregulares, com predomínio nas regiões peribrônquicas e cissurais, e adenomegalias mediastinais (hilares e paratraqueais). As alterações em geral são simétricas e ocorrem principalmente em regiões médias e superiores. A existência de sinais de fibrose pulmonar (faveolamento) indica doença em estágio IV. É comum a dissociação clínica e radiológica, isto é, extensa doença radiológica apresentando pouca repercussão clínica (oligossintomática e discretos achados no exame físico).
A avaliação inicial inclui exames laboratoriais (bioquímica, provas hepáticas, calcemia, calciúria e exame comum de urina), provas de função pulmonar, TCAR, exame oftalmológico, ECG e ecocardiograma (e, conforme resultados, Holter de 24 horas), reação de Mantoux e outros, conforme os achados clínicos. A espirometria pode apresentar resultado normal, de padrão restritivo ou obstrutivo, dependendo do tipo de comprometimento pulmonar. A capacidade de difusão em geral está reduzida, nos pacientes com envolvimento do parênquima, e pode estar diminuída em 20 a 40% naqueles em estágio I. Realiza-se o diagnóstico por meio de biópsia de um local afetado, a qual evidencia granuloma sarcoide (sem necrose caseosa, de padrão imunológico), associado à exclusão de outras doenças granulomatosas (beriliose, toxoplasmose, micoses, paraneoplasia, colagenoses, PH, tuberculose). Em casos de sarcoidose pulmonar, o material é obtido em geral por biópsia transbrônquica ou de gânglios mediastinais por mediastinoscopia. Quando realizado, o LBA evidencia linfocitose com predomínio de CD4 sobre CD8. Diferentemente da maioria dos pacientes com DPPDs, muitos indivíduos com sarcoidose permanecem assintomáticos ou apresentam remissão espontânea. Nos casos de progressão da doençaou de envolvimento sintomático significativo (pulmonar em estágio II ou II com sintomas e alterações funcionais significativas, ou extrapulmonar potencialmente grave, como sarcoidose cardíaca ou neurológica), há indicação de tratamento geralmente com corticoide. Em alguns casos, é necessário o uso de imunossupressor, como o metotrexato ou a leflunomida.
Figura 105.17
Pneumonite crônica causada por amiodarona.
Observam-se (a) Áreas de consolidação subpleural com retração do parênquima e (b) bronquiectasias de tração.
865
Figura 105.18
Estágios radiológicos da sarcoidose pulmonar.
Tanto a tuberculose miliar quanto o envolvimento extenso por tuberculose de disseminação endobrônquica evidenciam um comprometimento parenquimatoso difuso. Em pacientes com tuberculose miliar, há ruptura de um granuloma na corrente sanguínea, com disseminação hematogênica, que envolve os pulmões e os outros órgãos (medula óssea, baço, fígado, entre outros). O infiltrado pulmonar é de padrão miliar, isto é, os micronódulos são bem definidos com acometimento difuso (Fig. 105.19). Em casos de disseminação endobrônquica, o padrão é consolidativo heterogêneo e assimétrico, e muitas vezes pode ser identificada uma cavidade que origina o processo (Fig. 105.20). Os sintomas são tosse, dispneia, hemoptise, febre, sudorese e emagrecimento. Estabelece-se, diagnóstico por exame de escarro (espontâneo ou induzido) e, eventualmente, por LBA, e, em casos de tuberculose miliar, por biópsia pulmonar (geralmente por broncoscopia) ou de outro órgão afetado (fígado, medula). Realiza-se o tratamento com tuberculostáticos.
As vasculites pulmonares são doenças inflamatórias que afetam geralmente vasos de pequeno e médio calibre. As doenças mais comuns são a granulomatose de Wegener, a poliangeíte microscópica e a síndrome de Churg-Strauss. As manifestações podem incluir dispneia, tosse, hemoptise e sinusopatia, além de alterações sistêmicas (nefropatia, neuropatia periférica, lesões cutâneas). Pode ocorrer uma apresentação aguda com insuficiência respiratória. As alterações radiológicas são, entre outras, lesões cavitárias, infiltrados intersticiais evanescentes e comprometimento alveolar difuso (em geral por hemorragia alveolar). A dosagem de ANCA é utilizada para o diagnóstico e para o acompanhamento, sendo o padrão c-ANCA sugestivo de GW, e o p-ANCA de outras vasculites. Estabelece-se o diagnóstico por meio de biópsia pulmonar ou de outro sítio, em geral, renal. O tratamento envolve realização de corticoterapia e uso de imunossupressores, eventualmente plasmaférese.
Figura 105.19
(A) Tuberculose miliar. (B) Há infiltrado pulmonar difuso de padrão micronodular.
O quadro do paciente é de evolução crônica e sem imunossupressão conhecida. Os exames laboratoriais são inexpressivos (incluindo provas inflamatórias normais), exceto por FAN reagente (título de 1:80). A TC de tórax evidencia infiltrado intersticial de padrão reticular (espessamento de septos interlobulares e de cissuras), bronquiectasias de tração e áreas de faveolamento subpleural, áreas de vidro fosco nas bases pulmonares com cerca de 30% de extensão, inexistência de adenomegalias mediastinais e derrame pleural. A espirometria com broncodilatador e as medidas de volumes pulmonares apresentam restrição pulmonar moderada (CVF de 1,8 L, 54%, CPT de 3,2 L, 58%) e capacidade de difusão pulmonar com redução também moderada (DCO corrigida pela hemoglobina [DCOc] de 12 L, 44%). O paciente é submetido à broncoscopia com LBA (neutrocitose, citopatológico sem células malignas, pesquisas e culturas infecciosas sem germes) e à biópsia transbrônquica (alteração inflamatória inespecífica em parênquima pulmonar). Como o quadro tomográfico não é típico de FPI, o paciente é submetido também à biópsia pulmonar cirúrgica, a qual evidencia padrão de pneumonite intersticial usual, confirmando, então, o diagnóstico clínico, radiológico e patológico de FPI, já que não havia causa identificada para fibrose até o momento. Embora FAN reagente em títulos baixos possa ocorrer na população idosa ou com FPI, o paciente permanece em acompanhamento para uma possível colagenose oculta. Inicia-se tratamento com N-acetilcisteína, via oral (600 mg, três vezes ao dia), e inclui-se o paciente em um protocolo de pesquisa de uma nova medicação para FPI.
Figura 105.20
Tuberculose pulmonar de disseminação endobrônquica. Observam-se típicos nódulos centrolobulares e padrão de comprometimento bronquiolar de árvore em brotamento (setas).
1.Baugman RP, du Bois RM, editors. Diffuse lung disease. a practical approach. London: Arnold; 2004.
2.Nead MA, Morris DG. Interstitial luna disease: a clinical overview and general approach. In: Fishman AP, Elias JA, Fishman JA, Grippi MA, Senior RM, Pack AI, editors. Fishman’s pulmonary diseases and disorders.4th ed. New York: McGraw-Hill; 2008. p. 1105-24.
3.Raoof S, Amchentsev A, Vlahos I, Goud A, Naidich DP. Pictorial essay: multinodular disease: a high-resolution CT scan diagnostic algorithm. Chest. 2006;129(3):805-15.
4.Kairalla RA, Carvalho CRR. Fisiopatologia das doenças pulmonares restritivas. In: Carvalho CRR, editor. Fisiopatologia respiratória. São Paulo: Atheneu; 2005. p. 211-9.
5.Zeng-li W. Advances in understanding of idiopathic pulmonary fibrosis. Chin Med J. 2009;122:844-57.
6.Noble PW, Homer RJ. Idiopathic pulmonary fibrosis: new insights into pathogenesis. Clin Chest Med. 2004;25(4):749-58.
7.Leslie KO. Pathology of the idiopathic interstitial pneumonias. Exp Lung Res. 2005;31 Suppl 1:23-40.
8.Ryu JH, Daniels CE, Hartman TE, Yi ES. Diagnosis of interstitial lung disease. Mayo Clin Proc. 2007;82(8):976-86.
9.Menna Barreto SS, Fiterman J, Lima MA, organizadores. Sociedade Brasileira de Pneumologia e Tisiologia: manual de prática pneumológica. Rio de Janeiro: Guanabara Koogan; 2010.
10.Bagatin E, Kitamura S. História ocupacional. J Bras Pneumol. 2005;32 Supl 1:S12-S16.
11.Pereira CAC. doenças pulmonares parenquimatosas difusas. In: Menna Barreto SS, organizador. Pneumologia no consultório. Porto Alegre: Artmed; 2009. p. 335-64.
12.Silva CI, Churg A, Müller NL. Hypersensitivity pneumonitis: spectrum of high-resolution CT and pathologic findings. AJR Am J Roentgenol. 2007;188(2):334-44.
13.Raghu G, Brown KK. Interstitial lung disease: clinical evaluation and keys to an accurate diagnosis. Clin Chest Med. 2004;25(3):409-19.
14.Gazzana MB, Silva DR. doenças pulmonares parenquimatosas difusas. In: Xavier RM, Dora JM, Souza CFM, Barros E, organizadores. Laboratório na prática clínica: consulta rápida. 2. ed. Porto Alegre: Artmed; 2011. p.
15.Baughman RP, Culver DA, Judson MA. A concise review of pulmonary sarcoidosis. Am J Respir Crit Care Med. 2011;183(5):573-81.
16.Antoniou KM, Margaritopoulos G, Economidou F, Siafakas NM. Pivotal clinical dilemmas in colagen vascular disease associeted with interstitial lung involvment. Eur Respir J. 2009;33(4):882-96.
17.Hamzeh N. Sarcoidosis. Med Clin North Am. 2011;95(6):1223-34.
18.Fenoglio CM, Reboux G, Sudre B, Mercier M, Roussel S, Cordier JF, et al. Diagnostic value of serum preciptins to mould antigens in active hypersensitivity pneumonitis. Eur Respir J. 2007;29(4):706-12.
19.Bradley B, Branley HM, Egan JJ, Greaves MS, Hansell DM, Harrison NK, et al. Interstitial lung disease guideline: the British Thoracic Society in collaboration with the Thoracic Society of Australia and New Zealand and the Irish Thoracic Society. Thorax. 2008;63 Suppl 5:v1-58.
20.Silva CIS, Terra Filho M, Jasinowodolinski D, Muller NL. Atlas e diagnóstico diferencial: tomografia computadorizada de alta resolução do tórax Jorge Kavakama. Rio de Janeiro: Revinter; 2008.
21.Elicker B, Pereira CA, Webb R, Leslie KO. High-resolution computed tomography patterns of diffuse interstitial lung disease with clinical and pathological correlation. J Bras Pneumol. 2008;34(9):715-44.
22.Raghu G. Interstitial lung disease. In: Goldman L, Schafer AI, editors. Goldman’s Cecil medicine. 24th ed. Philadelphia: Elsevier; 2012.
23.Jawad H, Chung JH, Lynch DA, Newell JD Jr. Radiological approach to interstitial lung disease: a guide for the nonradiologist. Clin Chest Med.2012;33(1):11-26.
24.Reynolds HY. Present status of bronchoalveolar lavage in interstitial lung disease. Curr Opin Pulm Med. 2009;15(5):479-85.
25.Schwarz MI, King TE Jr, editors. Interstitial lung disease. 5th ed. Shelton: PMPH-USA; 2011.
26.Xaubet A, Ancochea J, Blanquer R, Montero C, Morell F, Rodríguez Becerra E, et al. Diagnóstico y tratamiento de las enfermedades pulmonares intersticiales difusas. Arch Bronconeumol. 2003;39:580-600.
27.Lynch JP 3rd, McCune WJ. Immunosuppressive and cytotoxic pharmacotherapy for pulmonary disorders. Am J Respir Crit Care Med.1997;155(2):395-420.
28.American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med. 2000;161(2 Pt 1):646-64.
29.Behr J, Demedts M, Buhl R, Costabel U, Dekhuijzen RP, Jansen HM, et al. Lung function in idiopathic pulmonary fibrosis: extended analyses of the IFIGENIA trial. Respir Res. 2009;10:101.
30.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis:evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788-824.
31.Tachikawa R, Tomii K, Ueda H, Nagata K, Nanjo S, Sakurai A, et al. Clinical features and outcome of acute exacerbation of interstitial pneumonia: collagen vascular diseases-related versus idiopathic. Respiration. 2012;83(1):20-7.
32.King TE Jr, Tooze JA, Schwarz MI, Brown KR, Cherniack RM. Predicting survival in idiopathic pulmonary fibrosis. Scoring system and survival model. Am J Respir Crit Care Med. 2001;164(7):1171-81.
33.Tzelepis GE, Toya SP, Moutsopoulos HM. Occult connective tissue diseases mimicking idiopathic interstitial pneumonias. Eur Respir J.2008;31(1):11-20.
34.Katzenstein AL, Myers JL. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med. 1998;157(4 Pt 1):1301-15.
35.Muller-Querheim J, Gaede KI, Fireman E, Zissel G. Diagnosis of chronic beryllium disease with cohorts of sarcoidosis patients. Eur Respir J.2006;27(6):1190-5.
36.Rossi SE, Erasmus JJ, McAdams HP, Sporn TA, Goodman PC. Pulmonary drug toxicity: radiologic and pathologic manifestations. Radiographics. 2000;20(5):1245-59.
37.Judson MA. The diagnosis of sarcoidosis. Clin Chest Med.2008;29(3):415-27.
38.Statement on sarcoidosis. Joint Statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999. Am J Respir Crit Care Med.1999;160(2):736-55.
Frankel SK, Jayne D. The pulmonary vasculitides. Clin Chest Med.2010;31(3):519-36.
Jayne D. Diagnosis of vasculitis. Best Pract Res Clin Rheumatol.2009;23(3):445-53.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.