(Carregando Índice)... (Carregando Índice)... |
Autor:
Jordana de Fraga Guimarães
Médica internista.
Última revisão: 26/05/2014
Comentários de assinantes: 0
Uma paciente do sexo feminino, 62 anos, branca, compareceu ao serviço de emergência devido a calafrios e febre com início há dois dias. Ela apresentou história clínica de síndrome mielodisplásica diagnosticada há dois anos. Naquele momento, uma anemia macrocítica com níveis normais de folato e vitamina B12 culminou em investigação adicional. A biópsia de medula óssea evidenciou uma medula hipercelular, com sideroblastos em anel e precursores eritroides multinucleados, e menos de 1% de blastos. A paciente foi tratada com azacitidina desde o diagnóstico. Ao realizar exame, ela estava febril (39,4oC) e taquicárdica (102 bpm) e apresentou pressão arterial de 110/70 mmHg e frequência respiratória de 22 rpm. Não foram obtidos achados significativos, no restante do exame físico. A partir do hemograma, verificaram-se hemoglobina de 7 g/dL, leucócitos de 42.000/ L e plaquetas de 36.000/ L. O esfregaço do sangue periférico mostra presença de bastões de Auer.
As síndromes mielodisplásicas constituem um grupo heterogêneo de distúrbios clonais adquiridos das células-tronco hematopoéticas, sendo caracterizadas por diminuição na contagem de uma ou mais séries hematológicas, hiperplasia medular e inúmeras alterações morfológicas e citogenéticas nas células hematológicas. Há risco de desenvolvimento de leucemia mieloide aguda (LMA) para pacientes com qualquer uma das formas de mielodisplasia. Essa é a condição hematológica mais frequente em idosos.
A mielodisplasia apresenta-se em duas circunstâncias clínicas distintas: primária (idiopática) ou relacionada à terapia. No primeiro grupo, a idade de acometimento é geralmente acima de 60 anos. A síndrome mielodisplásica relacionada à terapia ocorre 4 a 7 anos após o tratamento de neoplasias diversas com drogas quimioterápicas (p. ex., bussulfano, nitrosoureia, procarbazina, etc.) e/ou radioterapia. De 10 a 20% dos indivíduos tratados com essas modalidades terapêuticas podem ser afetados. O risco de desenvolvimento de LMA é maior com as síndromes mielodisplásicas relacionadas à terapia.
Muitos pacientes assintomáticos, até 50%, descobrem que são portadores da doença por meio de anormalidades evidenciadas no hemograma de rotina. A anemia é a principal alteração observada nesse exame. Quando sintomáticos, os sintomas e os sinais relatados são fadiga, dispneia, infecções de repetição e sangramento, os quais refletem insuficiência da hematopoiese medular.
Manifestações como febre e perda de peso sugerem mais um processo mieloproliferativo (p. ex., desenvolvimento de leucemia aguda) do que um mielodisplásico ou apresentam-se na fase tardia da mielodisplasia. As principais alterações verificadas no exame físico são palidez (anemia), púrpura e petéquias (trombocitopenia).
As principais causas de morte ocorrem devido a infecções (a mais comum) e sangramentos, que são complicações relacionadas à pancitopenia, e não à transformação leucêmica.
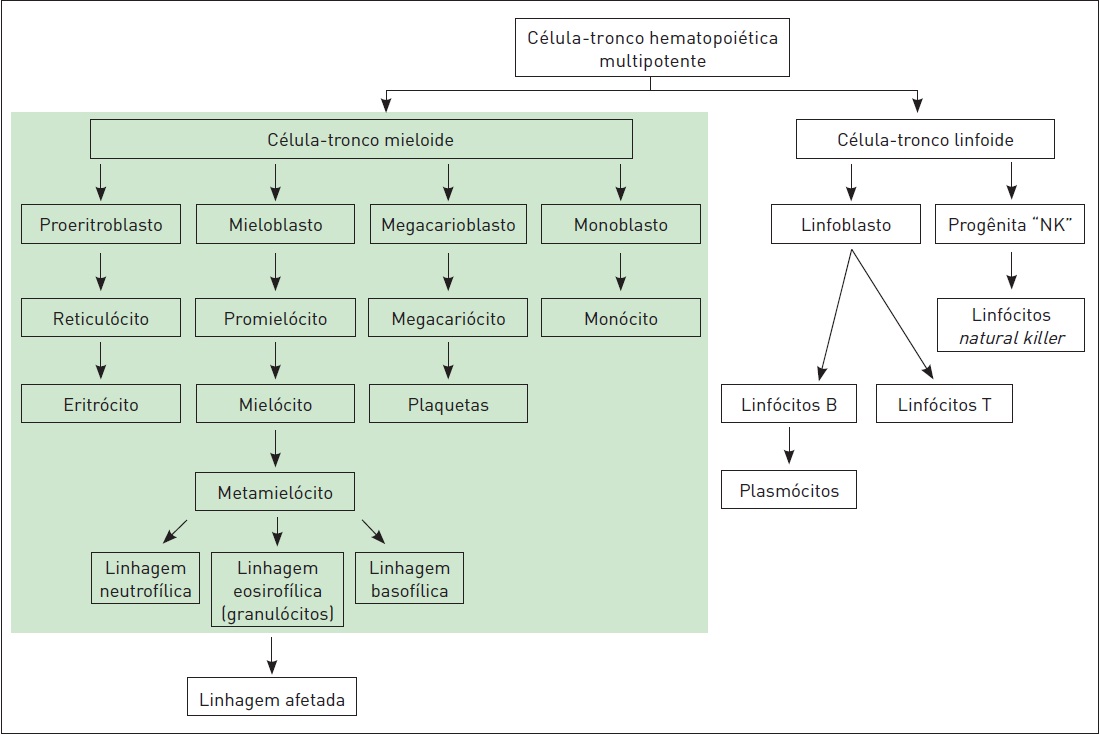
A medula óssea é parcial ou completamente substituída por uma progênie clonal multipotente (Quadro 57.1), que apresenta a capacidade de diferenciar-se em hemácias, granulócitos e plaquetas, porém de forma ineficiente. Em pacientes com síndromes mielodisplásicas, a maturação está defeituosa, e a hematopoiese é ineficaz – resultado de uma elevada taxa de apoptose. As anormalidades cromossômicas envolvem tipicamente os cromossomos 5 e 7, e a existência destas está associada a um pior prognóstico. A displasia afeta todas as linhagens não linfoides (eritroide, granulocítica, monocítica e megacariocítica) (Fig. 57.1).

Células-tronco
Existem várias plataformas de células-tronco que se diferem quanto à “potência”. Células pluripotentes (p. ex., células-tronco embrionárias) apresentam a capacidade de gerar todos os tecidos das três camadas embrionárias (ectoderma, endoderma e mesoderma). Células multipotentes (p. ex.,células-tronco de adultos) podem dar origem apenas a células especializadas de uma única camada embrionária (p. ex., células-tronco da medula que originam apenas células sanguíneas).
A anemia é o achado mais comum no hemograma de pacientes com síndromes mielodisplásicas, e esta pode estar associada a neutropenia e trombocitopenia. Essas duas alterações, isoladamente, não ocorrem com frequência. O volume corpuscular médio (VCM) apresenta-se normal ou elevado (macrocitose). A contagem de reticulócitos está normal ou baixa. No esfregaço de sangue periférico, macrovalócitos podem ser encontrados. Os neutrófilos são uma série frequentemente afetada, com neutropenia. As alterações morfológicas dos neutrófilos são redução da granulação, do número de lóbulos (pseudo-Pelger-Huet: neutrófilos com dois lóbulos) e existência de corpúsculos de Dohle. Formas jovens de células, como promielócitos e mieloblastos (em geral < 10%), podem ser encontrados também no sangue periférico. Havendo bastões de Auer em paciente com diagnóstico prévio de síndrome mielodisplásica, pode-se sugerir que esta já tenha progredido para LMA. A monocitose é um achado frequente nesses pacientes. O número de plaquetas pode estar reduzido, e as alterações morfológicas observadas são aumento do tamanho dessa célula e redução do número de grânulos.
O exame da medula é fundamental para a realização do diagnóstico. A medula óssea é, de forma característica, hipercelular, mas pode ser também normocelular e, menos comumente, hipocelular, em 20% dos casos, podendo causar equívoco de diagnóstico por aplasia de medula. Os indícios de eritropoiese anormal incluem características megaloblásticas, sideroblastos em anel e precursores eritroides multinucleados. O número de blastos está aumentado (essa quantidade correlaciona-se fortemente com o prognóstico), e os precursores granulocíticos são hipogranulares e hipossegmentados. Os megacariócitos apresentam uma redução no número de núcleos.
O risco de progressão para LMA depende do número de blastos na medula – quanto maior, mais elevado é o risco. Menos de 10% dos pacientes com a forma anemia refratária desenvolvem LMA. Já os pacientes com excesso de blastos ou leucemia mielomonocítica crônica (LMMC) evoluem para essa complicação em 20 a 50% dos casos e apresentam um prognóstico mais desfavorável (ver classificação adiante).
O principal diagnóstico diferencial das mielodisplasias deve ser realizado com LMA. Arbitrariamente, se há mais de 20% de blastos na medula, o diagnóstico é de LMA. A anemia megaloblástica também é um importante diagnóstico diferencial. Enquanto na mielodisplasia há neutrófilos com redução da lobulação, na anemia megaloblástica o aumento da lobulação é um fator característico.
Um pior prognóstico está relacionado a deleções completas nos cromossomos 5 e 7.
Figura 57.1
Fluxograma das linhagens eritroide, granulocítica, monocítica e células natural killer (NK) megacanocítica.
Fonte: Adaptada de Le e colaboradores.¹
Clinicamente, é de grande utilidade a classificação elaborada pelo French-American-British Cooperative Group2 para essas síndromes.
Classificação do French-American-British Cooperative Group:
Anemia refratária (AR, 21%) – < 5% de blastos na medula; < 1% de blastos no sangue periférico; < 15% de sideroblastos em anel na medula.
Anemia refratária com sideroblastos em anel (ARSA, 17%) – < 5% de blastos na medula; < 1% de blastos no sangue periférico; 15% de siberoblastos em anel na medula.
•Anemia refratária com excesso de blastos (AREB, 37%) – 5-19% de blastos na medula.
•Anemia refratária com excesso de blastos em transformação (AREB-t, 12%) – 20-30% de blastos na medula, > 5% de blastos no sangue periférico ou a presença de bastões de Auer.
•Leucemia mielomonocítica crônica (LMMC, 13%) – =20% de blastos na medula, < 5% de blastos no sangue periférico, monocitose > 1.000/mm3.
A Organização Mundial da Saúde, em 1999, propôs algumas modificações para essa classificação.3 Nessa nova classificação, a AREB-t e a LMMC são retiradas e classificadas como a primeira no grupo das LMA e a segunda no grupo das síndromes mielodisplásicas/ mieloproliferativa respectivamente. O grupo AREB foi subdividido em AREB-1 (< 10% de blastos) e AREB-2 (> 10% de blastos). Duas novas categorias foram criadas: citopenia refratária com displasia multilinear (CRDM) e citopenia refratária com displasia multilinear e sideroblastos em anel (CRDM/SA). Os pacientes com deleção 5q também foram separados em um grupo à parte.
O International Prognostic Scoring System (IPSS) é um escore prognóstico que considera a porcentagem de blastos na medula óssea, as alterações citogenéticas e a gravidade das citopenias.
Não há uma terapia efetiva para essas doenças. A única alternativa de cura para as síndromes mielodisplásicas é o transplante alogênico de células-tronco; no entanto, o papel dessa intervenção é limitado pela idade frequentemente elevada e pelo curso insidioso. A taxa de sobrevida é de 50% em três anos.
A administração de azacitidina é um tratamento efetivo para a melhora dos sintomas, da contagem de hemácias e para prolongar o tempo de conversão para LMA.
O uso de lenalidomida foi recentemente aprovado para o tratamento da anemia em pacientes com mielodisplasia e que necessitam de transfusões frequentes. A eficácia dessa terapia é particularmente alta nos indivíduos com deleção 5q.
Os pacientes com neutropenia primária grave podem utilizar fatores de crescimento mieloide, como o G-CSF. As medidas de suporte consistem na transfusão de hemácias e na administração de eritropoietina semanalmente para os pacientes com anemia grave. A ação da eritropoietina, em alguns indivíduos, aproximadamente 20%, reduz a necessidade de transfusões repetidamente.
O paciente, de 62 anos, apresenta diagnóstico prévio de síndrome mielodisplásica, com sintomas súbitos de febre e calafrios. Devido à febre, sugere-se mais um processo mieloproliferativo (p. ex., desenvolvimento de leucemia aguda) do que mielodisplásico apenas. Obviamente, qualquer paciente com história de síndrome mielodisplásica, que se apresente com febre, deve ser avaliado para verificar existência de sepse (deve-se considerar as anormalidades da contagem e/ou a função dos granulócitos que podem predispor às mais diversas infecções). Contudo, no caso desse paciente, a existência de bastões de Auer sugere transformação leucêmica.
1.Le T, Bhushan V, Tolles J. First Aid for the USMLE Step 1 2011. New York: McGraw-Hill; 2011.
2.Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gral- nick HR, et al. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol. 1982;51(2):189-99.
3.Harris NL, Jaffe ES, Diebold J, Flandrin G, Muller-Hermelink HK, Vardiman J, et al. The World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues. Report of the Clinical Advisory Committee meeting, Airlie House, Virginia, November 1997. Ann Oncol. 1999;10(12):1419-32.
Catenacci DVT, Schiller GJ. Myelodysplasic syndromes: a comprehensive review. Blood Rev. 2005;19(6):301-19.
Kasper D, Fauci A, Longo DL, Braunwald E, Hauser SL, Jameson JL, editores. editores. Harrison: medicina interna. 16. ed. Rio de Janeiro:McGraw-Hill; 2006.
McPhee SJ, Papadakis MA, Tierney LM, editors. Current: medical diagnosis and treatment. 47th ed. New York: McGraw-Hill; 2008.
Mufti GJ. Pathobiology, classification, and diagnosis of myelodysplastic syndrome. Best Pract Res Clin Haematol. 2004;17(4):543-57.
Zacharias DG, Nelson TJ, Mueller PS, Hook CC. The science and ethics of induced pluripotency: what will become of embryonic stem cells? Mayo Clin Proc. 2011;86(7):634-40.
O MedicinaNET é o maior portal médico em português. Reúne recursos indispensáveis e conteúdos de ponta contextualizados à realidade brasileira, sendo a melhor ferramenta de consulta para tomada de decisões rápidas e eficazes.
| Medicinanet Informações de Medicina S/A Cnpj: 11.012.848/0001-57 | info@medicinanet.com.br |
MedicinaNET - Todos os direitos reservados.
Em função da pandemia do Coronavírus informamos que não estaremos prestando atendimento telefônico temporariamente. Permanecemos com suporte aos nossos inscritos através do e-mail info@medicinanet.com.br.